
Some time ago I noticed that Mother Nature, despite its amazing virtues, does not know how to make C-N bonds by oxidation. Think about it: there are so many C-N bonds out there (in DNA, in alkaloids, you name it), yet none of them are made using oxidative enzyme machinery. If you are thinking about C-O bonds, then there are many examples of their construction using oxidative enzymes such as p450’s and others. However, all C-N bonds have reduction as their origin or are made using carbonyl condensation reactions. One might speculate as to why this might be the case. From Mother Nature’s standpoint, the most plausible reason would be “why bother?”. The metal-based nitrogen oxidants are inherently more difficult to produce compared to their oxygen counterparts (for example, metal nitrenes vs metal oxo species), so there is no evolutionary reason to go high in energy if the key bond-forming events can be accomplished using simpler means. The key factor is that molecular oxygen is our readily accessible terminal oxidant and there is just no nitrogen analog that is similarly abundant. We later wrote a short commentary in Nature Chemical Biology discussing this problem. This paper continues to be well cited (primarily by synthetic folks who need a believable justification for why synthetic nitrogen transfer systems are cool):
http://www.nature.com/nchembio/journal/v2/n6/abs/nchembio0606-284.html
Now it turns out that chemists can teach Mother Nature some new tricks. Take a look at the paper by Bollinger and co-workers in Nature Chemical Biology:
http://www.nature.com/nchembio/journal/v10/n3/full/nchembio.1438.html
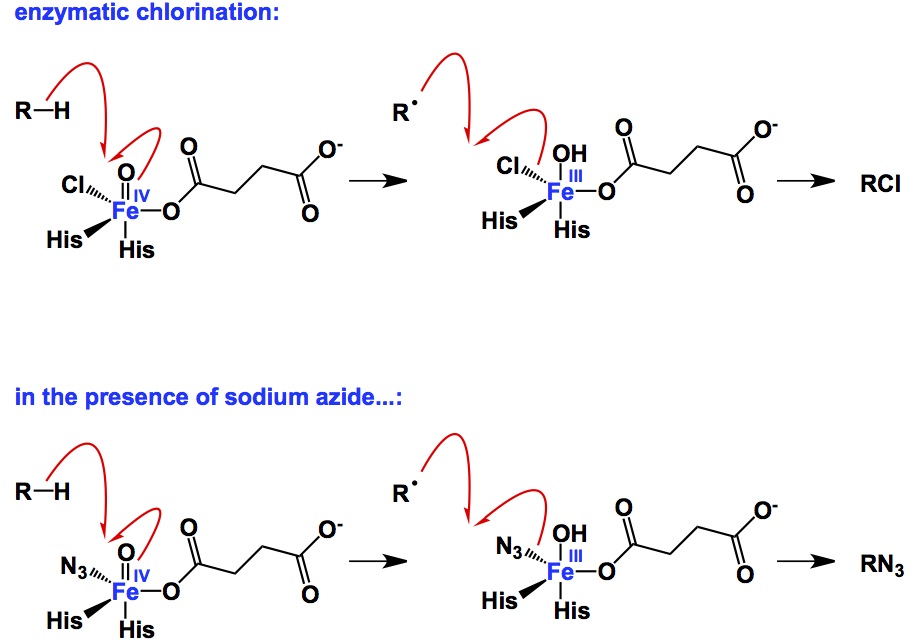
The α-ketoglutarate/iron-dependent dioxygenases and halogenases are typically recruited to run a wide range of enzymatic reactions ranging from hydroxylation to halogenation. Despite this useful palette of reactions, no C-N coupling by this class of enzymes has previously been reported. Bollinger et al. discovered that an αKG/Fe-dependent halogenase, SyrB2, can catalyze aliphatic nitration and azidation reactions. I am showing the azidation process above, which takes place when excess sodium azide is fed to the enzyme (thus, azide anions outcompete chloride anions). Despite the fact that chemical yields in the present version of the process are still very low, this study opens up new possibilities for nitrogen transfer by selective enzymatic C-H activation. It is conceivable that engineering of substrate specificity through directed evolution might be achieved in the future iterations of this system.
