We know that association does not necessarily mean causation. But does causation always imply association? This is trickier, yet situations like this are more common than you might think. I think that they severely complicate experimental science. In medicine this is commonly referred to as unanticipated confounders. This is why clinical trials need to be constructed with great care. A new medicine may have the intended effect, but the heterogeneous patient population can affect analysis when a pre-existing condition in a particular age group counteracts the marker you have chosen to measure the effect caused by treatment.
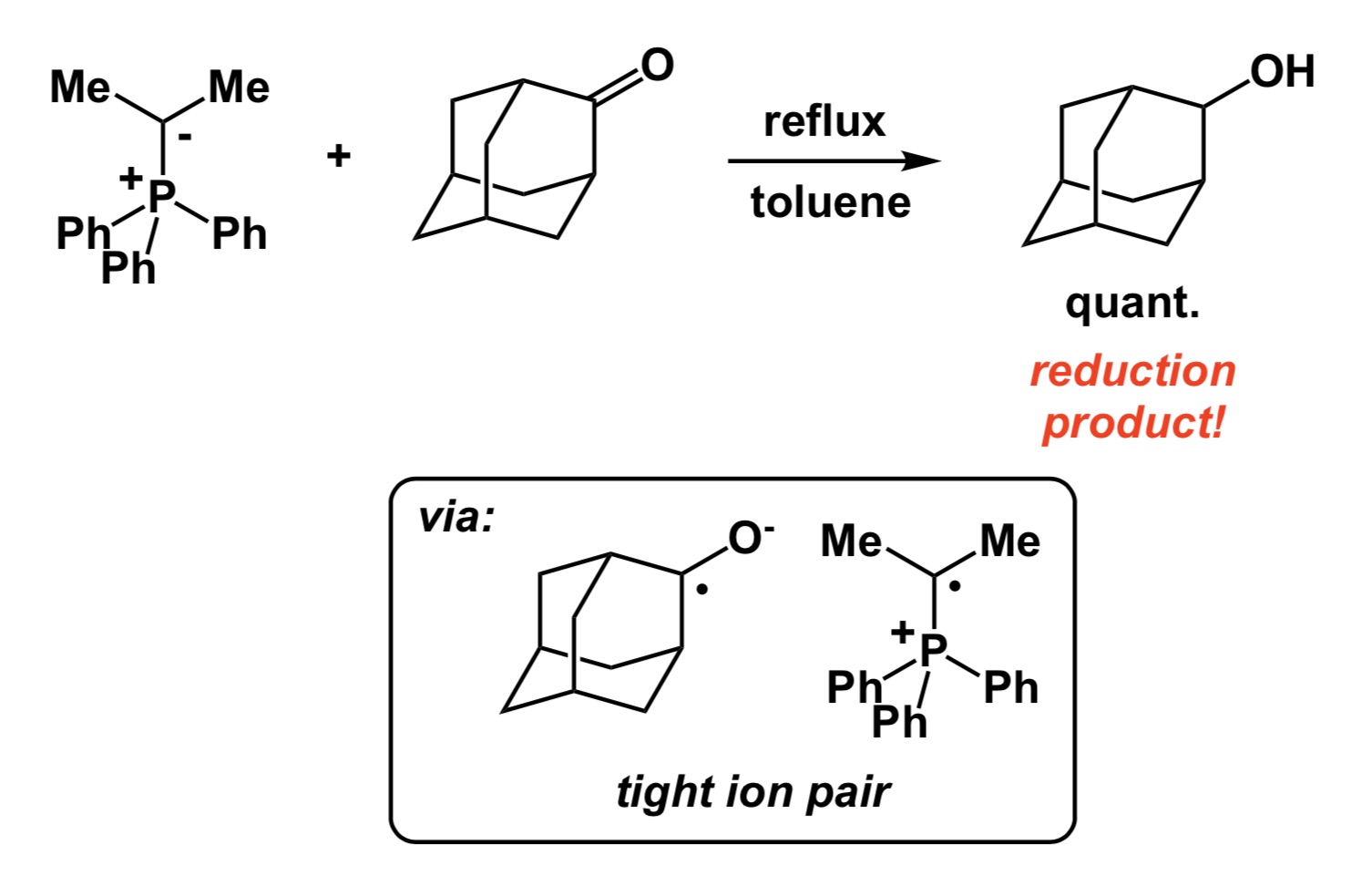
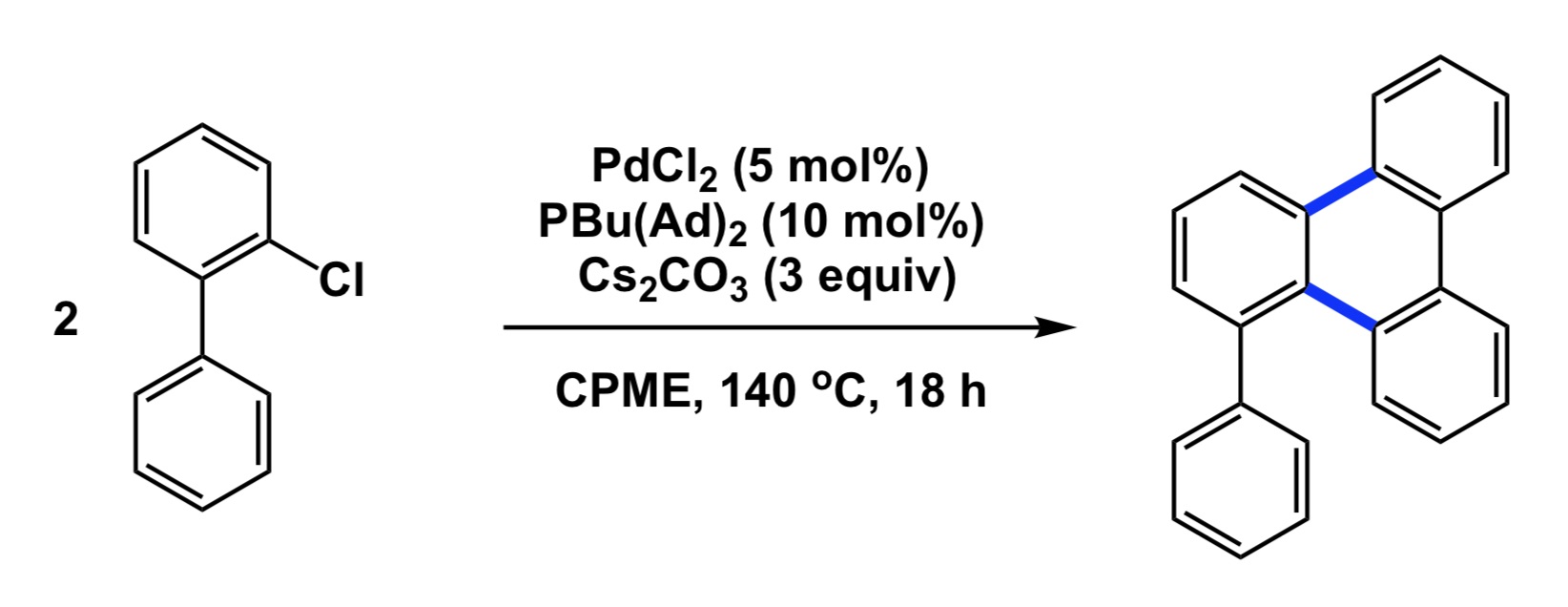
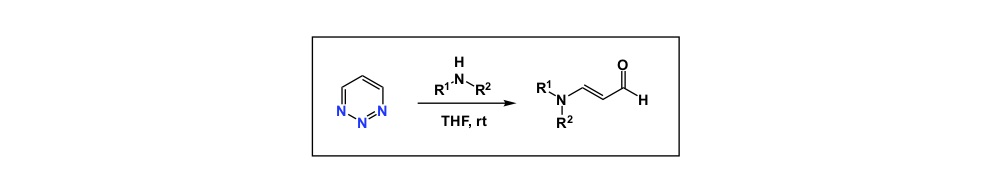
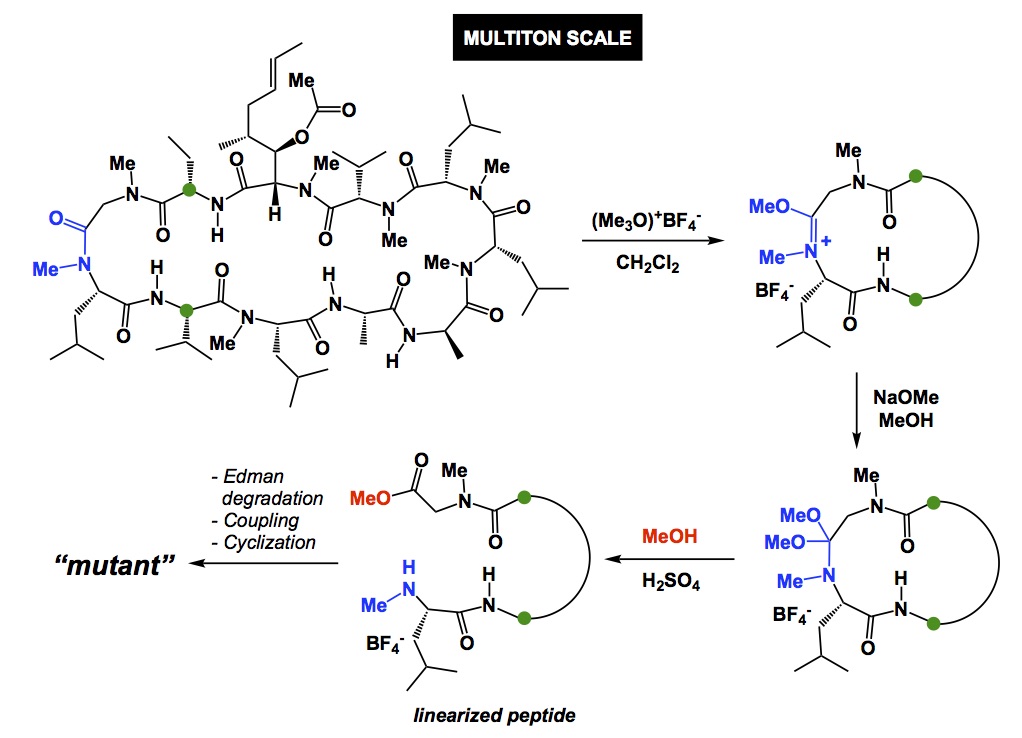
Now a chemistry example. If you mistakenly select a relatively electron-rich aromatic compound as internal standard to follow some catalytic oxidation, you may observe little conversion of your starting material and think that the new process you study does not have any value. However, your poorly chosen internal standard is sensitive to oxidation and if the rate of its degradation is similar to that of your substrate, you cannot possibly conclude that the new condition represents an improvement, yet it very well may be. So, causation may be there and you may have a superior oxidation process, but you have not established association.