
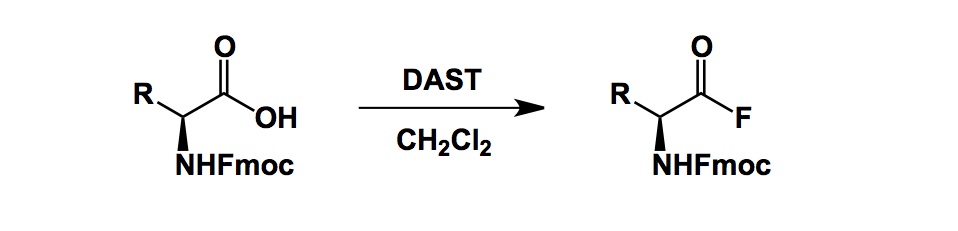
Earlier today, our Encycle team had a discussion regarding some of the issues pertaining to N-Me amino acid coupling reactions. Overall, we typically have no trouble introducing N-Me groups into peptides. All we need is an appropriately protected N-Me building block for the solid-phase Fmoc chemistry and off we go. The trouble is, sometimes this coupling fails miserably if hindered amino acids are brought together. There is an ingeneous solution to the problem and it offers a workaround. The reaction was developed by Professor Schafmeister of Temple University and it involves amino acid fluorides as electrophiles. A few words about them: in brief, acid fluorides are remarkably stable to aqueous hydrolysis. In contrast to their chloride congeners, fluorides also resist racemization. One of the classic ways to prepare them is through the use of DAST (there are better alternatives these days). Kaduk’s way cited below is as simple as it gets – just mix an Fmoc-protected amino acid with DAST in dichloromethane and get your acid fluoride product after aqueous work-up. The product can be crystallized from dichloromethane/hexanes, which makes for a rather practical method.
http://link.springer.com/article/10.1007%2FBF00142240#page-1
Back to the hindered amide workaround developed by Schafmeister. The mechanism involves mixed anhydride formation, which is the central trick here. Once this electrophilic intermediate has been formed, the rest is “downhill history” as nitrogen acylation becomes an intramolecular process. Even exceptionally hindered amides can be made using this procedure. Connoisseurs of multicomponent reaction might notice a similarity with Ugi’s mixed anhydride. We are attempting to run the Schafmeister process this week (wish us luck, we have a tough substrate that failed with everything else).
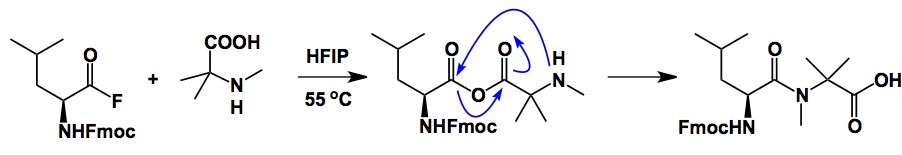

This is very nifty. By the way, if you are using acyl fluoride and make them in sit from TFFH, don’t buy the expensive reagent – it is very easy to make, in a two-step one-pot procedure, on a mol scale.
For the most hindered acylations in solution, or for amine partners that are very electron deficient, when everything else fails, try Fmoc-AA-chloride (pure, synthesized separately using oxaloyl chloride and dried carefully on highvac) + AgCN (4 equivalents) instead of a base, room temp to 60C, 1 hour in anhydrous DMAc or DMEU, (benzene also works as a solvent but is less clean), fairly concentrated (0.5-1M) reaction mix. The reaction is heterogennous throughout, protect from light by wrapping the flask in Al foil to avoid darkening, at the end cool. filter off the Ag salts, and rinse the solids with ethyl acetate, the usual aqueous extractive workup of filtrates follows. No detectable racemization. This obscure acylation protocol saved my bacon on more than one occasion.
Thanks for the trick. This will be relayed to our team asap! When you say “dried carefully”, I assume there was no heat involved, right?
right, just drying on highvac overnight to remove trapped oxalyl chloride (which was used in 1.2 equivs, in DCM, with one drop of DMF as a catalyst, at room temperature, for few hours after the gas evolution stopped, followed by evaporation). As you know Fmoc aminoacyl chlorides do not store well, it is best to make them and use them immediately within the next day.
The acylation protocol is based on this reference: Bull. Chem. Soc. Japan 49(8), 2335-6 (1976). The method is compatible with Fmoc protection. I replaced HMPA as a solvent with anhydrous DMEU (available from Fluka), later I saw that anhydrous DMAc also works well as a solvent, and is considerably less expensive.
Very interesting. Will check it out for sure!