
It is always exciting to witness when fragment-based approaches start from fairly mundane molecules and lead to potent inhibitors of enzyme function. By way of a reminder, in fragment-based drug discovery, screening of low molecular weight scaffolds is initiated with the goal to find weak, yet small, binders. The key is to locate scaffolds with high ligand efficiency (the change in free energy per non-hydrogen atom). The tools for measuring these intermolecular interactions range from protein/ligand co-crystallization to HSQC NMR. Progressive rounds of improvement lead to gradual transition to better and better molecules. A major limitation of this approach is when the inhibitor must induce substantial protein reorganization. Indeed, weakly binding fragments are not able to cause movement of large protein segments.
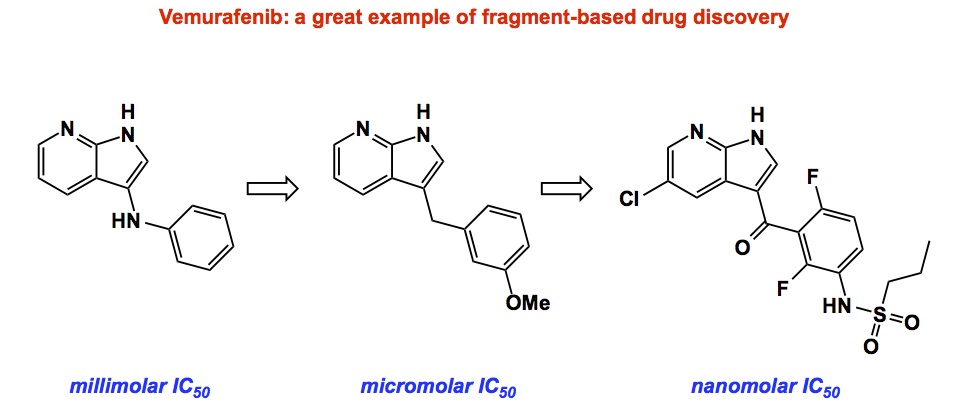
If you wonder whether or not fragment-based approaches have already resulted in drugs on the market, there are some notable examples out there. The most intriguing of them is that of Vemurafenib, the first drug approved for BRAF-mutant cancer. There is a great paper that details how this molecule was developed (http://www.nature.com/nrd/journal/v11/n11/full/nrd3847.html). Below I am showing a rough progression from the initially found small fragment to the larger, more “obese”, molecule. What’s interesting to me is that the process of gradual improvement did not center on the same kinase. If you follow the history of Vemuratenib, you will realize that the incremental improvement in potency corresponded to using different kinase isoforms at each step. This is an interesting twist that shows that fragment-based approaches are particularly well suited when you have many isoforms. I am not sure people stress this enough.
One of the protein targets we work on in collaboration with the SGC (Structural Genomics Consortium) has delivered some interesting hits. Our target is not a kinase (more on that some other time). We now have several compounds that show binding and co-crystallize with our protein. However, the proof for the effectiveness of this approach will come once we improve the initial hit, which has been problematic (we are still in the millimolar range). Honestly, it is somewhat disappointing to see a crystal structure with several good looking hydrogen bonds between your protein and its binder, only to be unpleasantly surprised by the low affinity of the interaction. I think if we had more target isoforms, similar to what the kinome offers, we would be able to move faster.
