
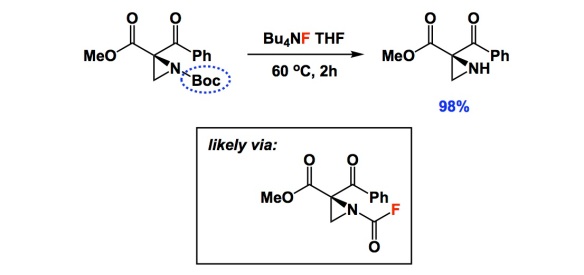
I typically do not comment on deprotection conditions, but there is something special in the two papers below. When I read the one by Lattanzi and colleagues, I thought that their nice asymmetric chemistry had been somewhat overshadowed by a single carbamate cleavage condition using TBAF. I am not sure how many of you are experienced with aziridines, but they do not easily withstand typical Boc removal with TFA. When I saw the yield of 98%, I was literally floored. In the interest of full disclosure, I learned about this deprotection from a paper I recently refereed. I dug a bit deeper and found that the TBAF condition goes back to the 2004 Tetrahedron report by Coudert and colleagues. In it, the authors considered a number of substrates and even carried out mechanistic studies that seem to suggest that the reaction proceeds through the formation of carbamoyl fluoride. Really strange, I know, but there is some very clever evidence in the Coudert paper. That 2004 study centered on the use of common amines and there was nothing as exotic as aziridines. Unless you are familiar with the pain of dealing with these three-membered rings, you might not think that a new way to remove Boc is worthy of note. This simple TBAF trick might be consequential to a lot of people interested in the chemistry of aziridines.
Lattanzi: http://pubs.acs.org/doi/abs/10.1021/ol3017066
Coudert: http://www.sciencedirect.com/science/article/pii/S0040402004012487

the trick is nice but I do not buy the mechanism with carbamoyl fluoride as intermediate. I was just reading the Coudet papers, and I think the carbamoyl fluoride is totally fanciful – they were using commercial 1M TBAF in THF and they forgot that commercial TBAF solutions are made from TBAF.3H2O. I am ready to bet 500 USD that the actual reagent is F(-).xH2O which acts as a milder version of HO(-)
1) If there were carbamoyl fluoride intermediate, we would see symmetric urea formation as byproducts. Sure enough Coudet et al saw them – but only in the specific case when they were deprotecting RNH-CO-OPh and they took it as a proof. But they have not observed symmetric ureas with any other carbamates they tried, e.g. with Boc. What they forgot was that phenolate is an excellent leaving group and active carbamates like RNH-CO-OAr are commonly used as more stable surrogates of isocyanates (they turn to RNCO under basic conditions, i.e. in TBAF.3H2O solution)
2) They describe fairly facile aminoacid ester hydrolysis with their refluxing TBAF system, and mention that ester hydrolysis buffers TBAF to a point it becomes ineffective – this again looks like OH(-) attack, not F(-). They see preferential Boc removal in the presence of ester only in case of highly activated Boc groups, such as 1-Boc indole, with a lazier aromatic acid ester, otherwise the ester is generally more reactive and is hydrolyzed first (BocTrpOMe provides BocTrpOH).
3. I think the observed selectivity of Boc to hydrolytic removal in the case of Boc aziridine ester is due to unique functional group arrangement – the two carbonyls are very close to the Boc group and the oxygen electrostatic repulsion is trying to force the Boc group carbonyl out of plane of the aziridine ring. We know from twisted amide analogy (e.g. 2-quinuclidinone) that twisting amide bond will greatly increase its lability towards hydrolysis.
There is simple way to test this – a) try selective Boc removal from aziridine with K2CO3 slurry in anh. MeOH b) replace TBAF.3H2O solution with anhydrous TBAF made in situ from Bu4NCN and C6F6
Well, as I mentioned: it is outlandish. In terms of my own initial reaction – I actually first suspected fluoride as base. To me, though, the urea experiment is interesting nonetheless. There are indeed other possibilities, as you point out. I, for one, would love to just see 19F NMR done in situ. Will be easy to detect the purported C(O)F thing…
it would be nice to see carbamoyl fluoride but the only way to preserve it (it will not survive reflux in wet THF-TBAF) is to use anhydrous TBAF and then the byproduct would have to be t-butoxide tetrabutylammonium salt, which I think it pretty unlikely. Dry TBAF solution will shut down the reaction. I think invoking fluoridolysis in this case is really not supported by available evidence.
This reminds me a case of KF supported on alumina. This is useful mild base is strong just enough to deprotonate phenols for alkylation, but mild to avoid epimerization alpha to carbonyls and some other base-promoted side reactions. For year, special effect of F(-) was invoked to explain its properties – Until someone really took time to characterize KF on alumina and found there were no active F(-) available (because of formation of insoluble AlF3). The basicity was due to potassium aluminate and simply combining alumina with KOH solution provided a supported base with nearly identical properties.
I recall Karl Christe (years ago) had a paper dedicated to making anhydrous TBAF. All it takes is isopropanol (several azeotropes) and then TBAF turns into this amazing white solid…
I think the route that uses Bu4NCN (which is easy to make and dry) with 0.2 eq. of C6F6 in THF is better – hexacyanobenzene is not problematic impurity and you form only 1/6 equiv of it. It is fast and you don’t have to worry about any protic impurity, and the obtained anh TBAF solutions store well
Thanks!
You were literally floored? I am picturing you peering forward at your computer screen and then being blasted off your seat by the realization
Man – you know it… This is the business we’ve chosen, when Boc deprotection knocks you off your feet. But I tell you what: had I known about this trick 10 years ago, we would have done many things differently. Not necessarily better, but differently.