
Years ago, when Iain Watson was in my lab, we looked at palladium-catalyzed allylic amination. We came to the conclusion that this reaction was far more nuanced than people had thought (http://pubs.acs.org/doi/abs/10.1021/ja055288c?journalCode=jacsat). Under acidic conditions, the process is under thermodynamic control and linear product originates from isomerization of kinetically formed branched allyl amine. The addition of base naturally suppresses this process and one can then isolate the branched derivative. I asked our grad students to identify the origins of this selectivity as part of a cumulative examination last December. I received some very reasonable proposals, but none was close to what is actually at play in this system. This is fine, but it exposes an interesting pedagogical challenge: people rarely turn to thermodynamic control as their first choice for explaining reaction outcomes. I suppose we are “wired” to seek uniquely distinct product-specific pathways and do not like to offer explanations that are based on a “path continuum” that is traversed differently according to conditions.
That was a longish prelude to an outstanding paper by Breinbauer and colleagues. My PhD student Frank Lee discussed this work at one of our weekly research updates. Here is another nuance attributable to palladium. What you see is an initial formation of the N-allylated heterocycle, which is followed by an aza-Calisen rearrangement. As a result, the reaction provides access to some interesting side chain-allylated products.
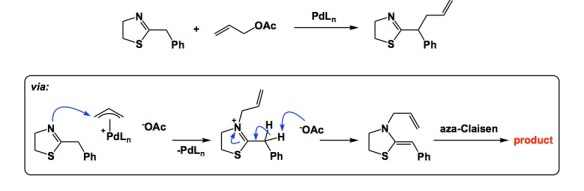

I am not an organic chemist and rarely read these type of publications. But, whenever I see Pd-catalyzed reactions, I am wondering if they isolated any dinuclear complexes. I hope someone writes an article about all the dinuclear complexes isolated in Pd-catalyzed reactions. That would be a great review article for people like me.
That is a great question. Modern catalysis has unveiled a number of cases in which polynuclear complexes are implicated. Purely organic papers, such as the one I mentioned, rarely go to that level of detail. But, as you know, absence of evidence is not evidence of absence.
Interestingly, we performed some experiments to determine the active Pd-species. Unfortunately, we were not able to isolate any crystals so far but we could exclude a dinuclear Pd-species as the active catalyst.
I can recomend some related publications regarding dinuclear complexes:
http://pubs.rsc.org/en/content/articlelanding/2016/cs/c5cs00537j#!divAbstract
http://pubs.acs.org/doi/full/10.1021/acscatal.6b02692
best wishes
MK
Thanks a lot! These are good papers indeed, will take a look a bit later. Great work.
regarding your isomerization during allylation – please was it amino group that was allylated? With a proton source around, Pd-catalyzed N-allylation becomes reversible, so I would not be surprised if with amine partially protonated the control was thermodynamic, as the more stable product eventually accumulates up to reaching the equilibrium level.
I guess that’s the reason behind using acidic allyl scavengers for amine deallylation, i.e. N,N’-dimethylbarbituric acid, plain non-acidic allyl sink would not work for the purpose.
The other thing I noticed, allylation with allyl acetates loves a primary carbon centers, kinetically, at least with plain old Pd(PPh3)4 as a catalyst. I have not done any kinetic studies but I noticed (with allylation of some piperazine NHs during a medchem project) that primary acetates and chlorides like allyl, cinnamyl were much faster than their secondary cousins.
So what happens is that the more substituted carbon is always attacked first, leading to the branched product. If you have acid, the amine is indeed protonated and this leads to re-formation of the allyl complex which now gives rise to the more thermodynamically stable linear product. This is what we found out. I recall that we chuckled at some computational papers (they appeared before our work), which explained why linear product was the preferred one on kinetic grounds. Having evidence against it made me wonder about the value of computation (it can explain anything, as you know).
sorry for my comment, only afterwards I had a look at your paper and then realized that you answered all the questions there, by doing very careful studies. I like your explanation of why aziridines are so deviant from other secondary cyclic amines. It would be interesting to include something like 2,2,2-trifluoroethyl amine, but the example with morpholine shows quite convincingly it is not just a pKa effect
Not a problem. By the way, it is hugely ironic that, while we know exactly what happened with other amines, to this day we have no idea why aziridine iss such an outlier.
interesting “amine” for comparison in allylation regioselectivity would be unsubstituted imidazole as nucleophilic partner. I would not be too surprised if it turned out to behave similar to aziridine – product forms under kinetic control, and there is no equilibration (even though alkylimidazoles are fairly basic and protonated imidazolium is a decent leaving group…)
Not sure we tried this, it has been a while…
I just got an idea for explaining the aziridine anomaly so that it becomes consistent with your other observations in the paper, and it predicts few additional things (i.e. 1,1-dimethylallyl N-substituted imidazole should not isomerize); I think there are some fine details to the equilibration mechanism proposed in the scheme 10 that would make the geometry more important than electronic factors and pKa: I actually believe the geometry on Pd(0) is fixed by aziridine ring strain to a point that Pd(0) becomes incapable of using allyl-substituted aziridine for allyl transfer whereas other amines including anilines are more accommodating to Pd(0) needs and have a very low barrier of isomerization as a result – but only if some H(+) is provided.
Please can I send you an e-mail to your chem.utoronto address? – I would like to avoid a public embarrassment if my proposal turns out to be dumb… Also, I would like to send a scheme in ChemDraw.
By all means, I will read! I have to say that Iain did look at these kinds of complexes as an explanation, this is a good lead, but there was still no concrete data (only computational…). I also suggest the following paper by us: http://pubs.acs.org/doi/abs/10.1021/ja076659n. Take away protons and it is all good (can get branched ones with “common” amines).
Thank you for the paper on DBU effect – it is a goldmine of information, and it fits neatly with your preceding JACS paper on isomerizations