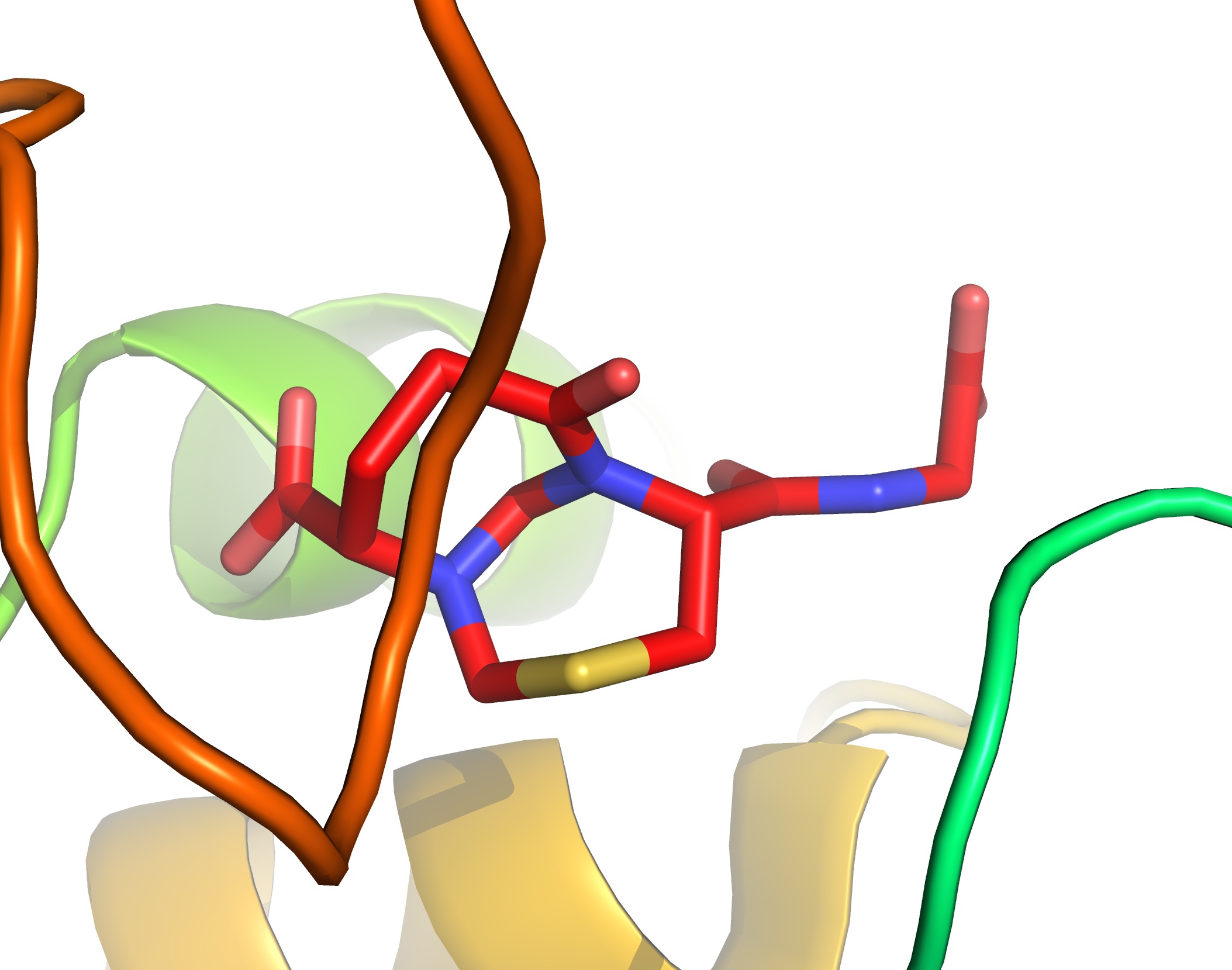
Xenobiotic electrophiles are “taken care of” by glutathione, which is part of our primary defence mechanism against various kinds of toxic molecules (this is why you do not want to take too many Tylenol tablets as the metabolite of this drug would quickly chew up your glutathione reserves, making you vulnerable). We do not often think about what happens to glutathione as a result of this chemistry. We probably think that it must be trivial and uninteresting. Certainly, the thiol group must play its key role in glutathione transformations, but I don’t think one tends to worry about the details of glutathione modifications. I was reading the 2007 paper by Kevan Shokat and colleagues in Organic and Biomolecular Chemistry and got reminded about the fascinating effects of formaldehyde, one of the simplest electrophiles, on polyfunctional molecules such as glutathione. Below you see a serendipitous finding made in the Shokat lab as part of a study aimed at non-enzymatic reactions between glutathione and formaldehyde. Not only did the insanely looking bicycle form, it also got co-crystallized with the carbonyl reductase I enzyme (pdb id: 2pfg). If you are interested in heterocyclic chemistry, this example of formaldehyde-driven construction of complex heterocycles should be of interest. I have always been fascinated by the increase in complexity that can be ascribed to the “stitching power” of this seemingly trivial molecule.

http://pubs.rsc.org/en/Content/ArticleLanding/2007/OB/b707602a