I concluded my productive trip to the University of Rochester yesterday, but not before I had a chat with Professor Damien Krysan. Damian has an interesting background, having worked at Abbott (medicinal chemistry), before doing an MD degree, which led to a faculty appointment at the University of Rochester. He is interested in antifungal agents and high-throughput screening, but this is not what crystallized my thoughts on the subject of today’s post. When Damian described his Abbott days, he reminded me of the early phases of using the AD (asymmetric dihydroxylation) process in industry. The development of the AD reaction was one of the truly exciting periods in the Sharpless lab and, although these developments predate my postdoctoral stay with Barry, I fondly remember some of the first-hand accounts of how the reaction evolved from the original pyridine effect defined by Criegee in the 1930’s (http://pubs.acs.org/doi/abs/10.1021/cr00032a009). The AD process acquired its rock-star status in the 1990’s, such that there was hardly an issue of JOC or JACS that did not contain an application of this technology in synthesis. The reaction was widely adopted in industry and, one might argue, ultimately defined many drug synthesis campaigns. This exemplifies a fascinating aspect of synthesis as it enables synergy between industrial needs and academic science. I want to make an argument that, in the ideal world, there must be a “perfect storm” of factors that lead to an explosion of interest in a particular synthetic process.
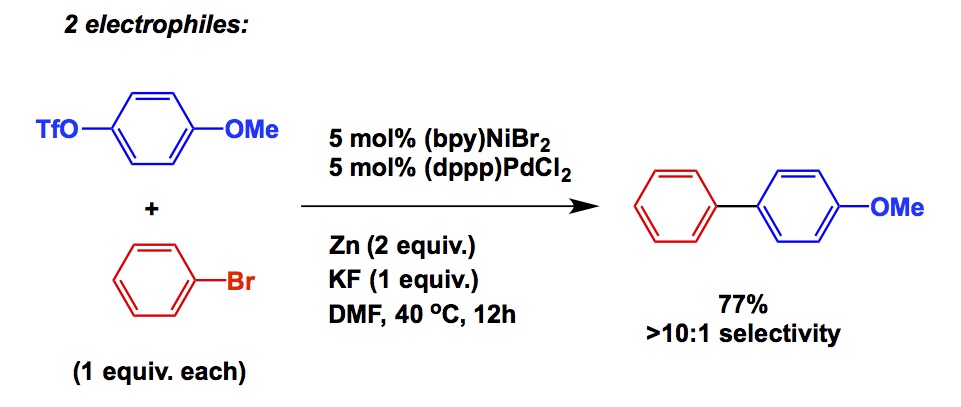
Just think about it: what were the biochemical targets at the very top of pharma’s wish list at the beginning of 1990’s? Undoubtedly, many of them were HIV proteases. Logically, sp3-righ frameworks decorated with vicinal heteroatom arrangements can be seen in a ton of protease inhibitor structures. This helped to popularize the AD process as it opened a clear path to molecules of that type. You do not see comparable levels of utility of this reaction nowadays, do you? This is because pharma has moved on and targets of today differ substantially from those of yesterday. As a corollary to that, one of the best things a synthetic chemist might want to do is anticipate the upcoming biological targets. I do not think anyone does this sort of stuff, but I can see how the knowledge of relevant chemotypes that tend to hit promising biological targets can only help those who want to stay ahead of the curve and make their chemistry widely used. I think that metal-catalyzed amination of aryl halides is another great example. Here we have a reaction whose development coincided with an explosion of interest in kinase inhibitors. Having said that, there was no biological “foreshadowing” of the fundamental studies by Buchwald and Hartwig (and I am not saying that there should have been one – it all worked out perfectly anyways).
Of course, an equally fascinating suggestion is that a simple-to-run process might influence the direction of drug discovery in industry. Indeed, synthetic chemists who work in pharmaceutical companies emerge from academia with unique preferences (there is a reason that Lipitor contains a pyrrole ring: Bruce Roth, the discoverer of this cholesterol-lowering agent, worked on pyrrole chemistry as part of his PhD). You might then say that it is a pity that reaction developers rarely care about biological relevance of their work and are not necessarily attuned to the latest trends of drug industry. However, perhaps none of this makes sense any longer because pnenotypic screens tend to drive industry as opposed to target-driven work. I don’t know.