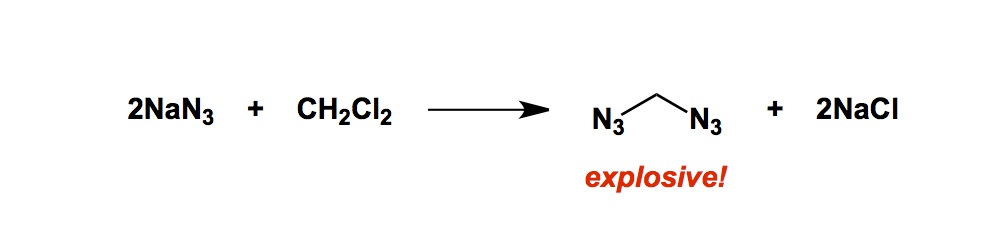
I am on my way to Europe for a week, which means that I will have less of a chance to update my blog. I will be on vacation with my wife, attending a wedding in Serbia, which will be followed by a couple of lectures in Germany – at Sanofi-Aventis and at the University of Mainz. There are many things one needs to do before going on vacation. There is always a ton of emails to write and several loose ends to tie up (crap, I just remembered about another one…). Lab safety is the item of substance that never leaves one’s mind before leaving town. Of particular concern is anything that is remotely dangerous and, in this regard, I have to admit that there are chemicals I admire much less than others. One of them is sodium azide and I just want to remind everyone not to use dichloromethane when dealing with this reagent. You would be surprised how many times I have encountered students in my lab who accidentally forgot this and turned to dichloromethane in conjunction with sodium azide. We all know how bad this combo is, yet somehow we forget. Why? Because we are the creatures of habit and dichloromethane is one of our trusted old friends in reaction work-up. Below is what might happen, though. I am also providing you a link to a paper that describes the explosion caused by diazidomethane. I am providing this link so that you do not think that my warning is purely anecdotal…