Last December, Dr. Steve Ritter of the Chemical and Engineering News asked me to comment on a paper from the lab of Prof. Petr Beier of the Czech Academy of Sciences. I gladly did (http://cen.acs.org/articles/94/web/2016/12/Fluorinated-azides-click-make-triazoles.html?type=paidArticleContent) and I just want to share my thoughts with you in the event you have not seen the Beier paper.
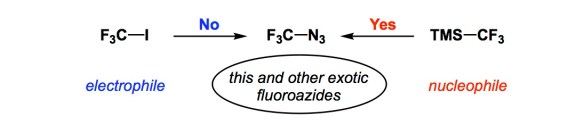
Every now and then we need a reminder of a rather straightforward kind: if we have trouble making a bond, just reverse the darn polarity of reagents! It is remarkable how infrequently this way of thinking pops into our heads, and I am judging from years of experience. Indeed, unless you are into radical reactions, there are always at least two ways to make a bond by a polar mechanism. In the example described by the Prague team in the Angewandte article, the curious CF3N3 molecule was the target. Attempts to forge the C-N bond using CF3I as the electrophile led nowhere, whereas using the fluoroalkyl portion as the nucleophile delivered CF3N3 and other uncommon azides without a glitch. I know this stuff ultimately relates to the well-known umpolung arguments, but those of us who are in the business of making bonds would still rather search “closer to the lamp post” than reverse reagent polarity. I am convinced that there are a lot of other previously “unmakable” molecules that might be made using this simple logic. We should keep this in mind.
http://onlinelibrary.wiley.com/doi/10.1002/ange.201609715/abstract
 Jokes aside, there is always a counterargument, isn’t there? Here is one in defence of our species: constrained by the lack of a universal energy carrier, we had no choice but to resort to these “intermediary” products to move electrons around. This is also a reasonable thought, ladies and gentlemen. Capital investment in electrochemical plants is not a joke and some of the arguments for direct use of certain kinds of energy (for instance sun light everyone talks about) eventually run out of steam because it is simply more economically feasible to have a carrier, however imperfect it might be. I always remember Surya Prakash’s dictum: there is no problem with energy on this planet, but there is a problem with the energy carrier. So we might be doing our best, after all.
Jokes aside, there is always a counterargument, isn’t there? Here is one in defence of our species: constrained by the lack of a universal energy carrier, we had no choice but to resort to these “intermediary” products to move electrons around. This is also a reasonable thought, ladies and gentlemen. Capital investment in electrochemical plants is not a joke and some of the arguments for direct use of certain kinds of energy (for instance sun light everyone talks about) eventually run out of steam because it is simply more economically feasible to have a carrier, however imperfect it might be. I always remember Surya Prakash’s dictum: there is no problem with energy on this planet, but there is a problem with the energy carrier. So we might be doing our best, after all.