The notion of privileged scaffolds originated in the pharmaceutical industry. The term relates to disproportionately high occurrence of a given scaffold among drugs and other bioactive molecules. One excellent review dealing with this subject comes from Snyder and Stockwell:
http://www.sciencedirect.com/science/article/pii/S1367593110000232
Piperazine is a well-known example of a privileged structure. The are two troubles with this presumably worthwhile status: a. it is tough to carve out a good intellectual property niche because there are just too many molecules that contain a privileged scaffold (by definition) and b. many privileged scaffolds are associated with promiscuous binding to their biological targets. Thus, you might not only have trouble obtaining good claims in your patent, but you might also have to deal with molecules that lack target selectivity. The piperazine case is actually not that bad, but a mere mention of a urea to someone in drug discovery might make him/her cringe.

Personally, I get annoyed when those who do synthetic methodology talk about a new method of making a certain sub-structure and tout the method’s significance because there is a lot of molecules out there that contain that structural element. If you are interested in discovering new bioactive molecules, this prevalence should be a good reason for NOT making the darn thing, but I let go of this argument when I talk to students who get excited about producing a sub-structure that is present in 3 thousand other molecules. Why spoil the party? After all, we are in the business of education and there is a sense of synthetic accomplishment if you make a molecule that contains some omnipresent core, particularly if there is an interesting mechanism involved. We even mention this in our own papers…
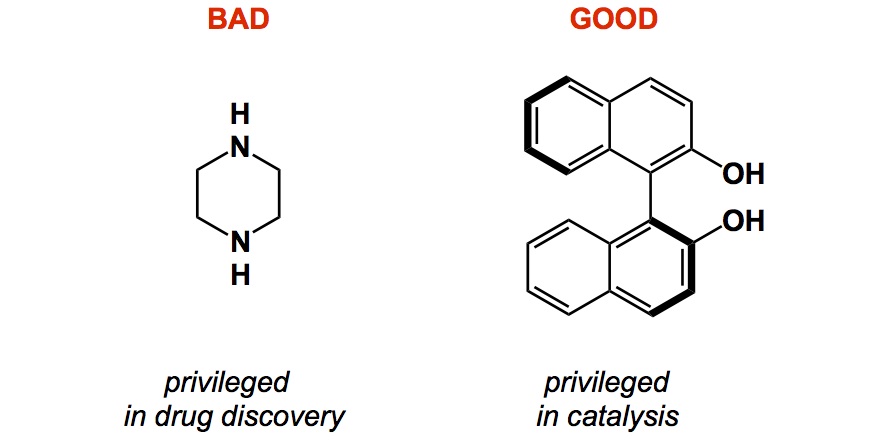
There are other kinds of privileged structures that extend beyond drug discovery. I think Eric Jacobsen was the first to bring this term to asymmetric catalysis (see the link below). Here, there are some wonderful molecules that indeed possess seemingly magical properties in terms of enabling better control of chiral space around a given catalytic centre. BINOL is a good example. You might argue that some researchers used to over-interpret the significance of C2 symmetry in relation to its catalytic prowess, but, without a doubt, in the case of BINOL we have a really good reason to call it a privileged core.
http://www.sciencemag.org/content/299/5613/1691.short
I suppose there is some irony in all of this. The term “privileged” came from drug discovery, yet I think there are major negative connotations to the “privileged” status of many bioactive structures, which is certainly not the case for catalysis, where we do not judge molecules on the basis of their interaction with proteins and DNA. As a result, there are some highly desirable attributes in a select few cores.