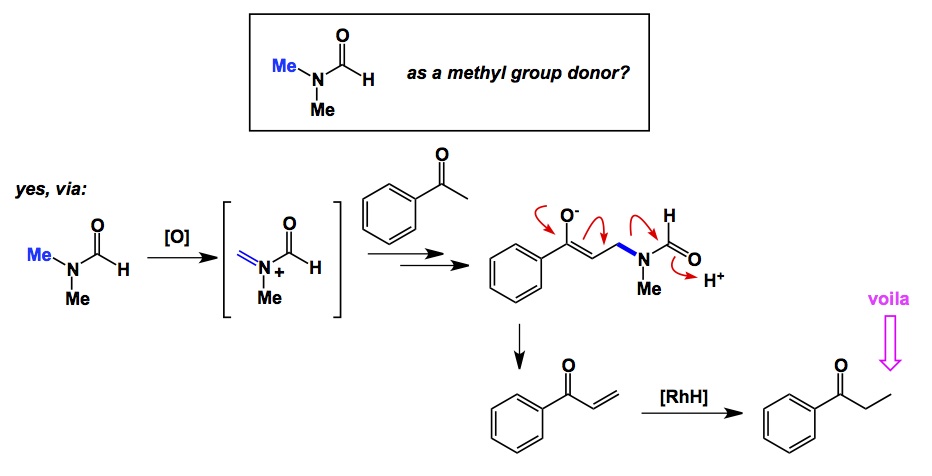
Where do some of the more obscure functional groups get their names? This is the topic for tonight’s discussion. For instance, everyone is familiar with amidines – you get them by replacing oxygen atoms with nitrogens in carboxylic acids. The properties change rather drastically, but I am not talking about them tonight. In fact, I will not even go into a protracted discussion about where amidines got their name from. There is a certain logical connection to amides here, and I am just going to leave it there. Now let’s switch the letters “m” and “d” in the name “amidine”. We are going to end up with adimines. Who are they? Maybe this name hints at some imine character? Well, despite the fact that I cannot, for the life of me, figure out the origins of adimines, these intermediates are absolutely fascinating, if rare. Take a look at the sequence shown below. The arylpyridinium salt is first hit with a hydrazine, followed by ring-closure to generate the adimine skeleton. In this particular work, courtesy of Alvarez-Builla’s lab, adimine serves as a springboard into other heterocycles by way of palladium catalysis. The reactions are interesting, especially to me since I am very fond of unusual nitrogen arrangements (here we have a all-sp2 NCNN sequence, which is really rare).

http://pubs.acs.org/doi/pdf/10.1021/jo801549u
In regards to names, I recall Nicos Petasis’s story of how his 5-year old daughter (at the time) corrected his mistake when she thought he misspelled “aminal”. Of course, she thought it must be “animal”…