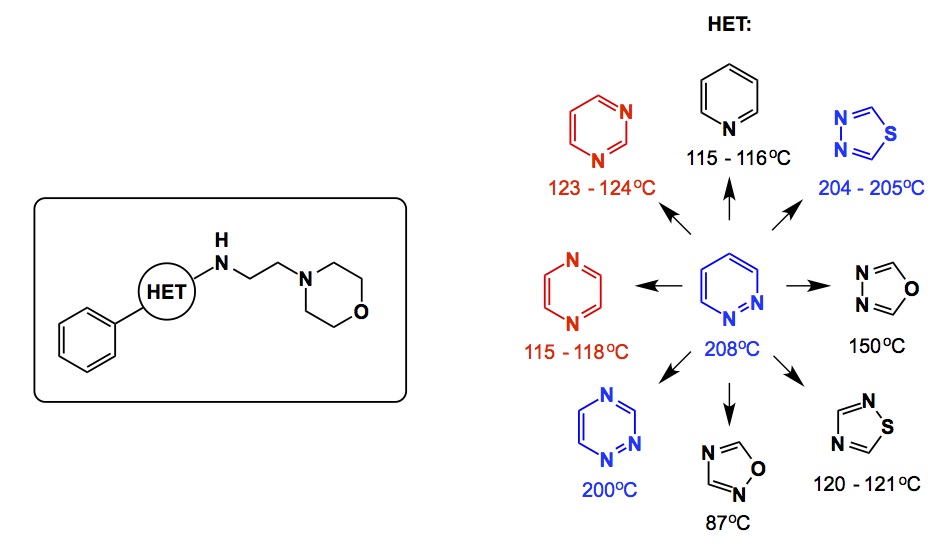
Imagine a bioactive compound about to undergo a medchem-style optimization. Let’s say this molecule contains a heterocyclic core. The question is: are there any measurable metrics that might suggest a rationale for replacing the existing core with some other heterocycle? In other words, do we have something in our disposal which is less Edisonian than “screen baby, screen”? I was really intrigued by a statement made by Wermuth in his MedChemComm paper. This article praises the virtues of pyridazines in medicinal chemistry (I am not going into that whole “privileged” business…). It turns out that for the particular biological application considered in this reference (biology is beyond the point tonight), the most potent cousins of the pyridazine-containing compound contained thiadiazole and 1,2,4-diazine rings, which are curiously close in their boiling points. The less potent derivatives contain pyrimidine and pyrazine, which also boil close to each other. The lesson here is that a boiling point reflects a dipole moment, which is an excellent comparative metric.
http://pubs.rsc.org/en/Content/ArticleLanding/2011/MD/c1md00074h#!divAbstract
This paper also reminded me about an awesome equation you can see below. This simple math relates melting point to lipophilicity and aqueous solubility. I think this is a great way to estimate solubility of molecules for which the melting point and lipophilicity data are available.