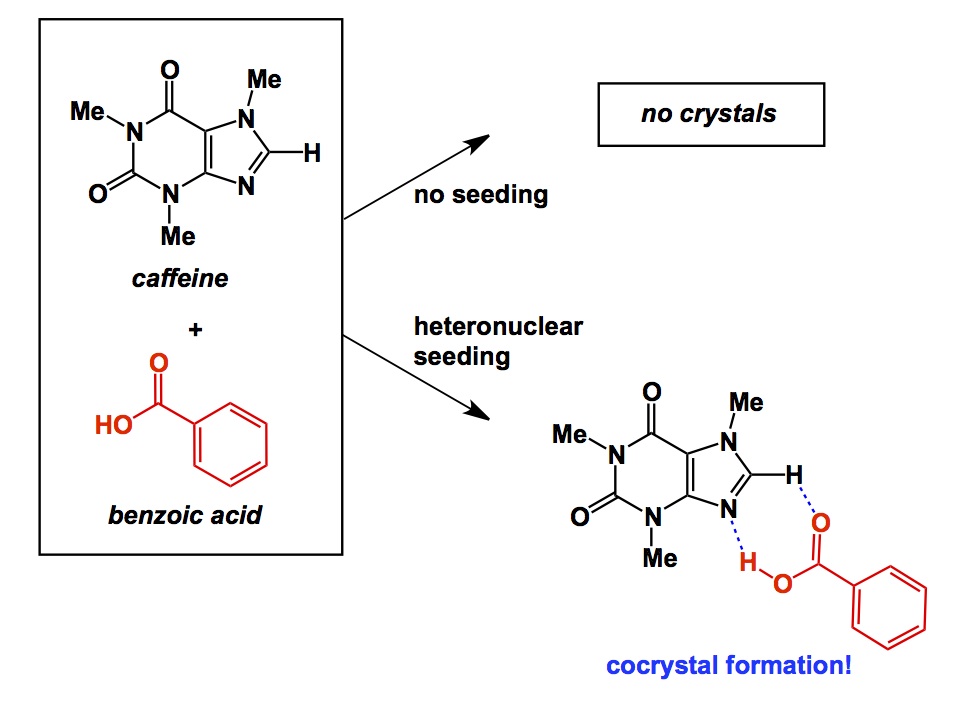
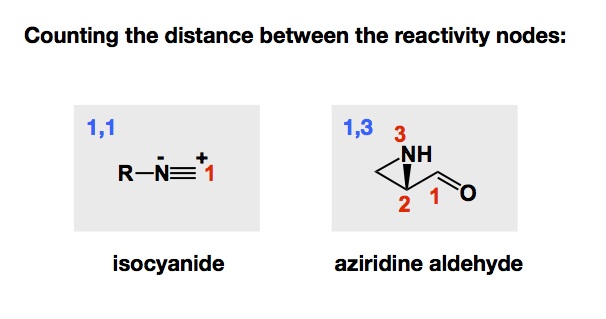
We have been having some exciting results in heterocyclic fragment screening using X-ray crystallography. The initial hits made with the help of Aman and Elena now enable further design and we will hopefully nail some of our targets together with Shinya, Conor, Jeff, and Rebecca in my lab. By “targets” I mean a series of methyllysine-binding proteins for which we intend to identify specific and cell-permeable probes. It will take us a while (longer than the remaining 2 months of my sabbatical), but we are in this thing for the long haul.
I want to cover one particular line of reasoning that has been brewing in my head for a while now. The sheer number of new (from my lab’s perspective) tools that have emerged out of the SGC collaboration, is quite staggering. They range from sophisticated soaking methods to co-crystallization and docking studies. I have turned into a total fan of Schrodinger Glide software (www.schrodinger.com). Dr. Conor Scully, a research associate in my lab, introduced me to this tool and I have continued learning on my own. I am having a lot of fun, although I sometimes feel bad about peppering the great Schrodinger team of developers with all sorts of questions. It is a full assault, I must admit, but these guys are real pros. Earlier today, Hege Beard, my contact there, informed me that she has developed an algorithm that will help us with covalent docking of aziridines to cysteine-containing protein targets. We have had some glitches with this, but the method is working really well now.
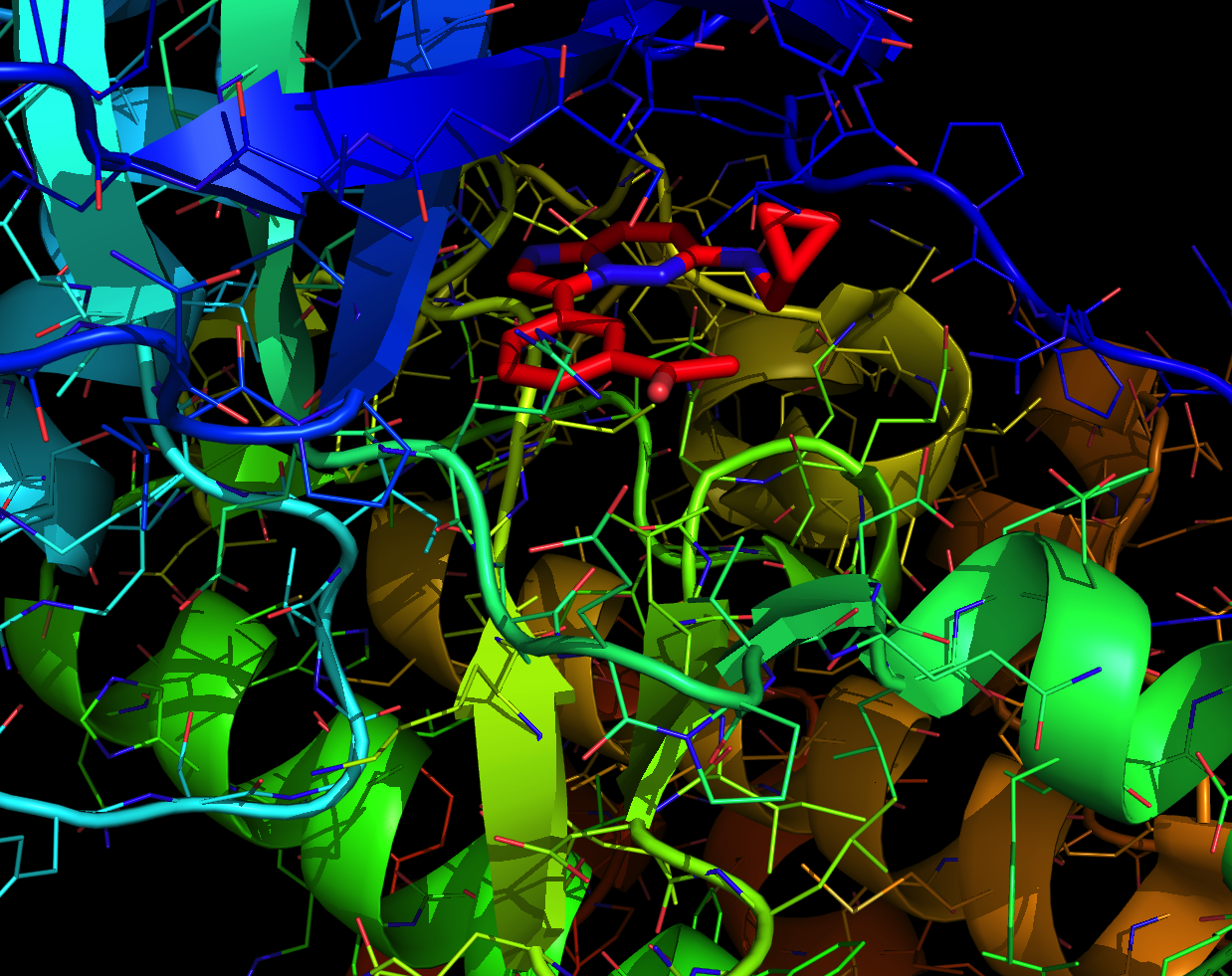
So where is my point for today, you might ask? Here it is. As I immerse into the elements of design and docking of ligands for our targets, I am forced to revise some of the dogmatic thinking I have been using in the past. For instance, one of the points I used to love chuckling about had to do with some curious publications that described the design of a molecule and experimental validation of its activity in biochemical and cellular assays. This would eventually lead to an atomic level view of the pivotal interaction, manifested in a co-crystal structure. And there came the moment of truth when scientists would humbly admit that their molecule, while active, potent, etc, engaged its target in a totally different way from their initial prediction. To back up my words with a concrete example, here is something form SGC. In this nice work, a kinase inhibitor (in red below) of Pim1 kinase was developed. I made this picture using PyMol. The co-crystal structure shows that the molecule does NOT bind in the anticipated “Type 1” mode (it is when the so-called hinge region is engaged in protein/ligand contacts).

http://cancerres.aacrjournals.org/content/67/14/6916.long
Having said that… I was recently on a lecture tour in B.C. and talked to a biochemist who told me about the so-called two-stage binding in cytochromes. This is totally unrelated and I am not an expert in this at all, but it makes me wonder about my previous position I just described. Here is my proposal: when you see a binding mode that is not consistent with the projected (designed) one, it may be a simple manifestation of kinetic/thermodynamic control and the fact that there are different phases to an interaction. In other words, what eventually crystallizes corresponds to the most stable arrangement that, by its nature, does not invalidate the design that took a different part of the binding site into account!