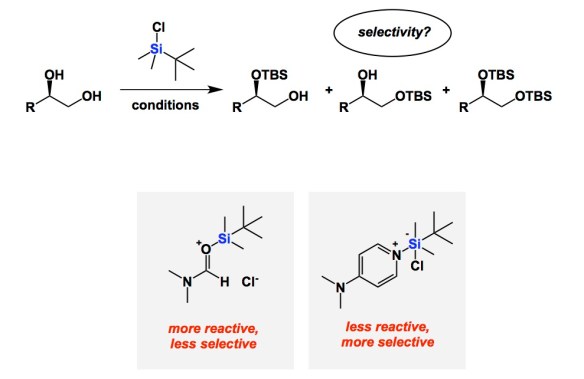
On a recent trip to Munich, I got acquainted with the research of Professor Hendrik Zipse. His mechanistic understanding of catalysis of alcohol silylation is both deep and educational. It serves as an instructive reminder of the reactivity/selectivity principle, which is something chemists relate to on an intuitive level. Not too long ago, Zipse and co-workers published a series of papers aimed at understanding the fundamental underpinnings of Corey’s classical silylation of alcohols. Zipse reminds us that DMF was the prescribed solvent in the original system. In this process, imidazole was used to mop up HCl, making TBS transfer one of the most familiar processes in organic chemistry. The question is whether or not the role of all components is crystal clear. It is now, but only after Zipse’s kinetic analysis. In brief, DMF is not your innocent by-stander. Its role is to form the active silylating argent, which is the Lewis acid/base pair shown below. Due to the high activity of this adduct, reactions in DMF (the original solvent from Corey’s 1972 paper) do not show impressive selectivity among primary, secondary, and tertiary alcohols. This is a very important finding. In contrast, if one stays away from DMF/imidazole mixture and runs silylations in dichloromethane along with DMAP and triethylamine, the selectivity is excellent. Improved reaction profile correlates with lower activity of the DMAP-derived active silyl transfer agent.