The pervasive dogma that complex problems require complex solutions is the root cause of many failed research programs. It is refreshing to hear when a complex phenomenon can be reduced to a limited set of parameters (preferably to just one). This does not happen every day and, when it does, I feel as if there is a breath of fresh air.
Let’s talk about protein-protein interactions. A lot of people are interested in developing small molecules that disrupt them, but I don’t think people know a lot about factors that drive the on/off rates when two protein partners engage each other. Most people (myself included) consider this to be an exceptionally difficult problem.
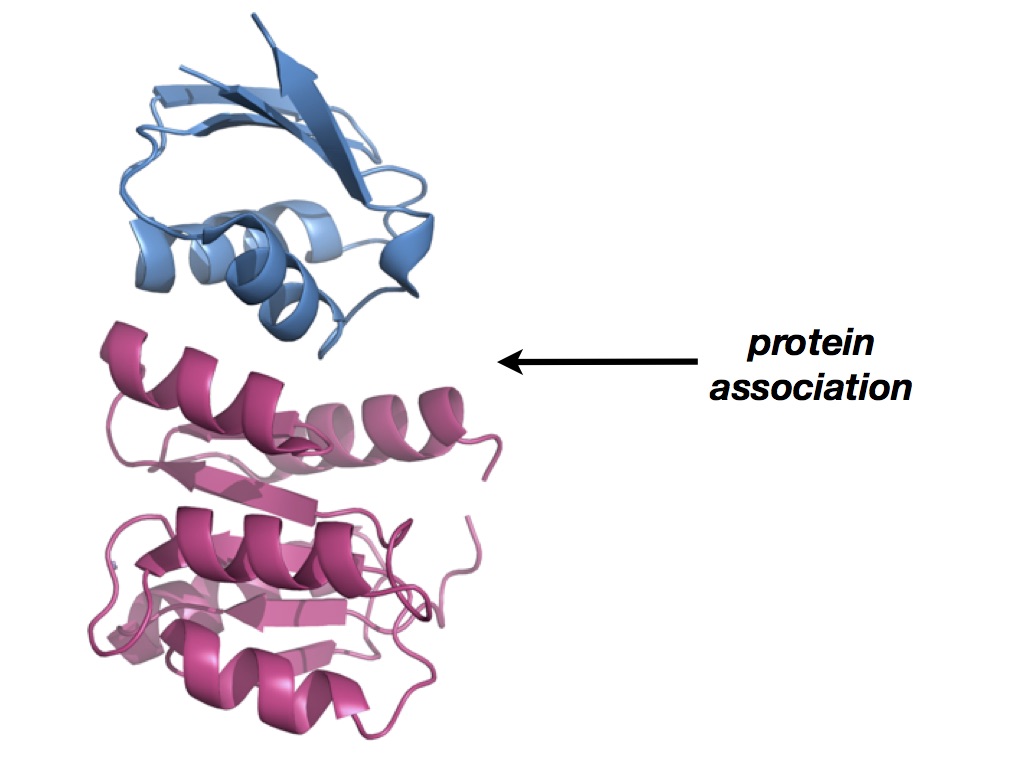
There is a paper by Das and Basu in Proteins that takes a remarkably simple view on the on/off rate problem. Believe it or not, the cosine of the angle between the dipole moment of one protein and the dipole moment of its interacting partner correlates strongly with experimentally determined on-rates, which measure protein-protein association constants. This finding was made upon analysis of 72 crystal structures and experimentally determined binding data. It will be interesting to see how this correlative analysis can be used to guide the development of protein-protein interaction inhibitors. There has to be a way of using the knowledge of electrostatic fields to one’s advantage (I have no idea about it, though).
http://onlinelibrary.wiley.com/doi/10.1002/prot.24860/abstract