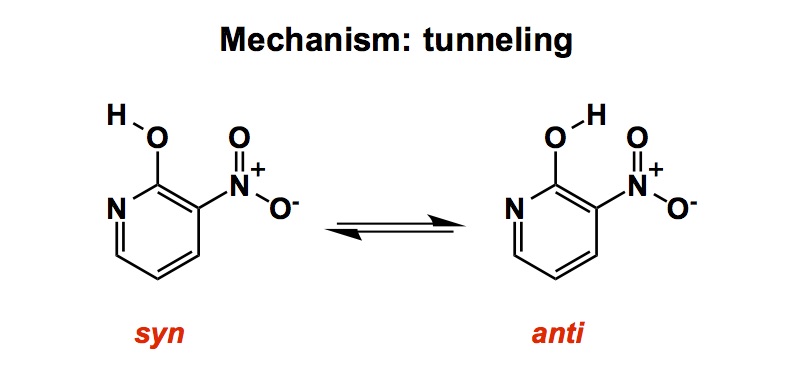
I don’t know about you, but sometimes all I want out of reading papers is pure basic science. I get particularly upbeat when I see an occasional outlandish structure that reminds me that not all ground states are created equal. Truth be told, with fewer and fewer people doing “blue sky” research, it is difficult to find such examples in the sea of utility-oriented manuscripts. While it is easy to get inundated with the amount of information being published, TOC (Table of Contents) graphics offer a glimpse into what to expect in a given paper. These devices were introduced by the publishers only about 13-14 years ago, which sounds crazy given how indispensible they seem to be. The trouble is that some of the most interesting vignettes and detours hardly ever appear in these graphics. As a result, it is virtually impossible to find real gems unless you read the whole thing (and who has the time for that?). Here is a good example: based on the TOC graphic alone, I could have easily missed some of the fascinating details in the paper from Hosoya and co-workers that appeared in Org. Lett. not too long ago. Upon perusing the contents of the article, I saw one of the characterized by-products. To me, this happens to be one of the most interesting results in this contribution.