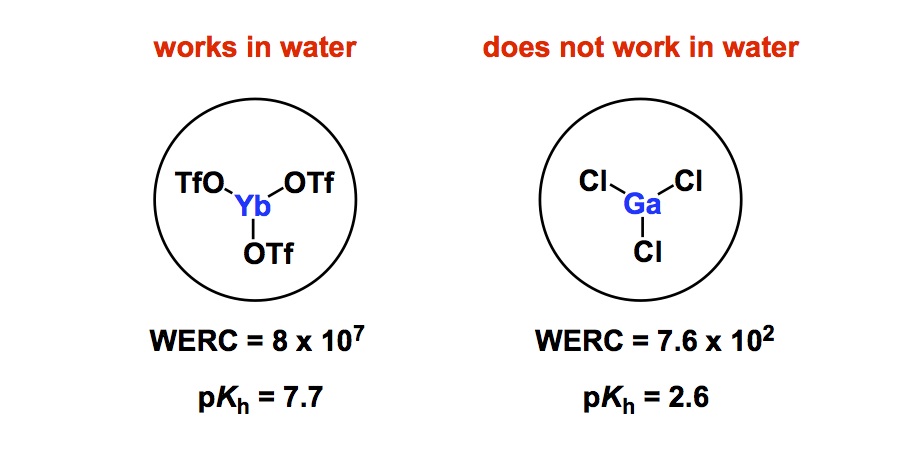
I want to talk about some unexpected and counterintuitive findings that run against what we might anticipate as chemists: that the introduction of a highly electronegative fluorine atom into a molecule necessarily increases the H-bond properties of adjacent functional groups. I refer to the work by Linclau and co-workers, which was published in Angewandte a couple of years ago. The cyclohexane-bound hydroxyl group was investigated in this study. The focus was on H-bond property of the OH functionality, which was measured using a really nice method: by looking at the decrease in absorbance of the hydroxyl substituent’s IR stretching band upon complexation with N-methylpyrrolidinone (NMP). Very cool stuff. Importantly, the paper casts doubt on the assumption that fluorination always increases H-bond acidity. There is a simple explanation of the observed effect – just take a look at the two representative examples shown below.

http://onlinelibrary.wiley.com/doi/10.1002/anie.201202059/abstract
The main lesson here is that fluorination can attenuate alcohol H-bond acidity in unanticipated ways. The intramolecular F···HO interaction can in fact be responsible for a decrease in H-bond acidity. Clearly, this intramolecular interaction can effectively outcompete the electron-withdrawing effect of fluorine which is expected to lead to increased acidity of the adjacent polar groups. The Linclau study opens doors for rational modification of acidity through site-selective fluorination and should have many applications in the design of bioactive molecules.