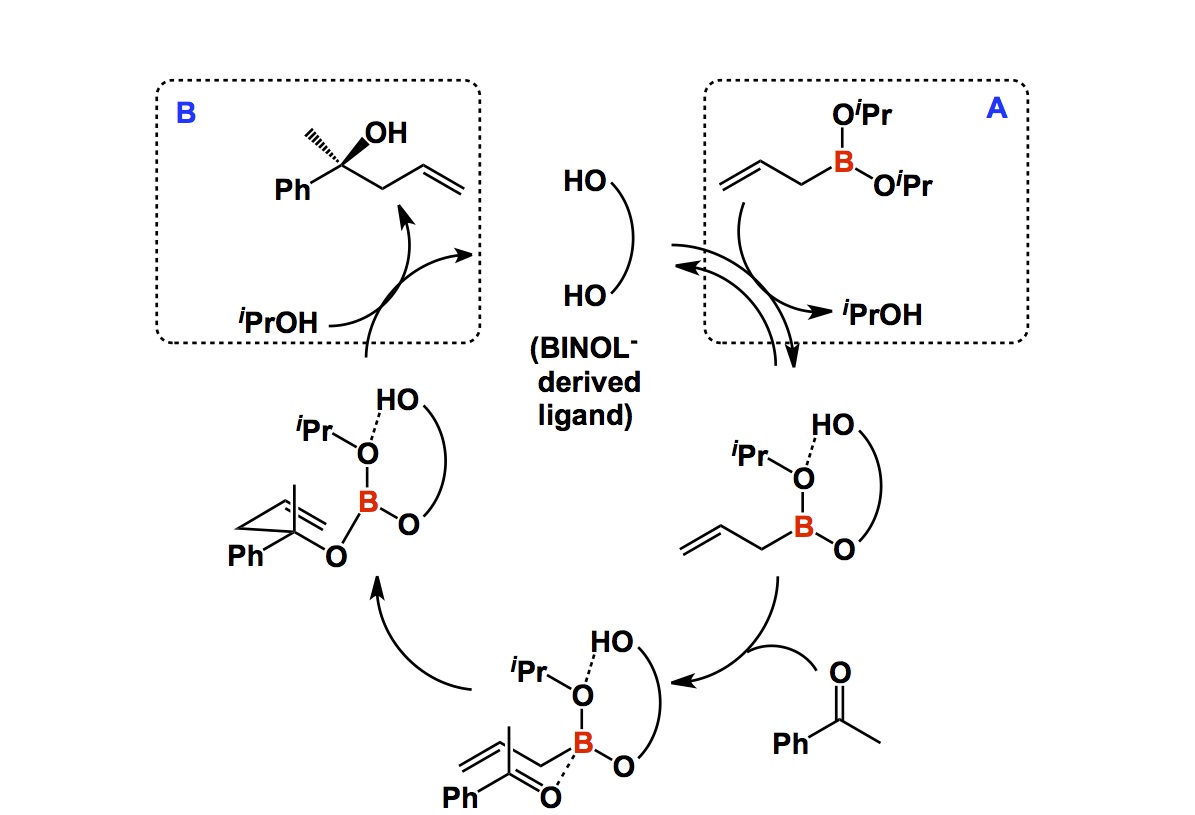
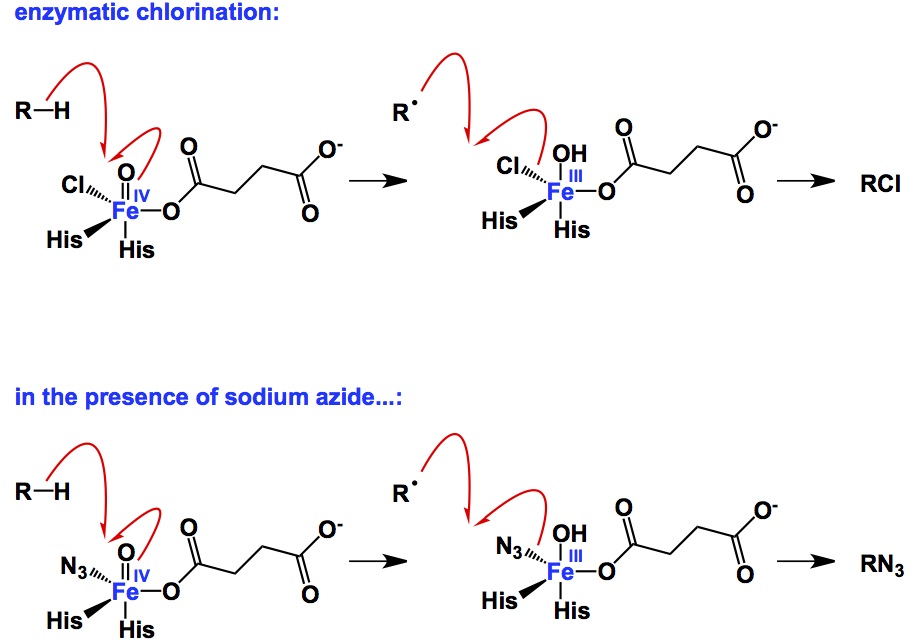
Aromatic heterocycles form the backbone of drug discovery. It is difficult to deny this statement for two simple reasons: a. the relative resistance of aromatic heterocycles to oxidation and b. their capacity to partake in a gamut of interactions with protein targets (hydrogen bonds, hydrophobic interactions, etc). While linking heterocycles into oligomeric chains is best done by way of cross-coupling reactions, there is no better alternative to condensations when it comes to making heterocycles themselves. Copper-catalyzed azide/alkyne cycloaddition is an exception to this rule. If you are thinking about a pyrrole, a pyridine, or a pyrimidine (the list can go on and on), nothing comes close to gaining aromaticity by kicking out water molecule(s) from a carbonyl precursor. Aromatic heterocycles that contain N-N or N-O bonds belong to a particularly vast class of useful molecules. Some time ago, I wondered about reactions that provide access to pyrazoles or isoxazoles by building a heteroatom-heteroatom bond as part of the process. For the life of me, I could not think of an example. You might say: why bother? As a matter of fact, I would agree because hydrazines and hydroxylamines are some of the most versatile and readily accessible nucleophiles. However, if I put my basic scientist hat on, I want to see reactions of this kind. Until we get there, my claim stays put: there are no examples where heteroatom-heteroatom bonds are made in the course of aromatic heterocycle synthesis.

http://pubs.acs.org/doi/abs/10.1021/ol801506y
I was reading a cool paper by the Swedish group led by F. Almquist and, upon a cursory look at one of the schemes, I said to myself: “Darn, this must be it! The N-N bond construction…”. Take a look above. On a sober glance, however, the reaction amounts to a Sandmeyer process gone “haywire”. In this reaction, the targeted diazonium intermediate activates the proximal methyl group. The reaction is rather unusual, which is why I like it. Still, this does not affect my assertion that there are no useful ways of making aromatic heterocycles by building heteroatom-heteroatom bonds. There might be something I am missing, of course. But I do not mean an obscure example, ladies and gentlemen. Please give me something synthetically useful.
Apart from the interesting pyrazole-forming reaction, this paper provides a neat example of peptidomimetic design. The tricyclic pyrazole-2-pyridone-thiazoline structures accessible with the Almquist method incorporate a dipeptide sequence within a rigid framework. Importantly, the two substituents that correspond to amino acid side chains may be varied, enabling construction of compounds libraries.