I was thinking about this subject for a while, not the least because my mother suffers from Alzheimer’s. In this disease, aggregation of amyloid-β peptides is widely thought to play a key pathological role. It is clear that prevention of the amyloid-β plaque formation would prevent memory loss. This strategy represents a potential for therapeutic intervention through a disease-modifying mechanism. A question arises regarding the biochemical path to the formation of amyloid-β peptides. Several enzymes contribute to the build-up of plaques. Of these enzymes, gamma- and beta-secretases have received a significant amount of attention from the research community. The enthusiasm in the early days of this research was grounded in the idea that inhibitors of gamma- and beta-secretases might block the production of amyloid-β plaques. Recently, however, gamma-secretase research has suffered a serious blow because a phase III clinical trial testing the inhibitor semagacestat failed (http://www.sciencedirect.com/science/article/pii/S009286741401304X). This led to closure of promising investigations aimed at inhibiting gamma-secretase and leaves behind beta-amino secretase as the best currently available target to prevent the formation of amyloid-β peptides. A seminal study by Stefansson et al. recently identified a missense mutation (A673T) in the amyloid-β precursor protein (APP) gene that protects against Alzheimer’s disease (http://www.nature.com/nature/journal/v488/n7409/full/nature11283.html). The mutation was identified by the analysis of the genome sequence data of 1795 Icelanders. I am truly amazed by the enabling features offered by the genetic makeup of this unique population. Interestingly, the A673T missense mutation reduces cleavage of APP by the beta-secretase BACE1. This finding supports the hypothesis that abnormal processing of APP causes Alzheimer’s disease. Significantly, the Stefansson paper further validates BACE1 as the legitimate target in search of treatment against Alzheimer’s disease.
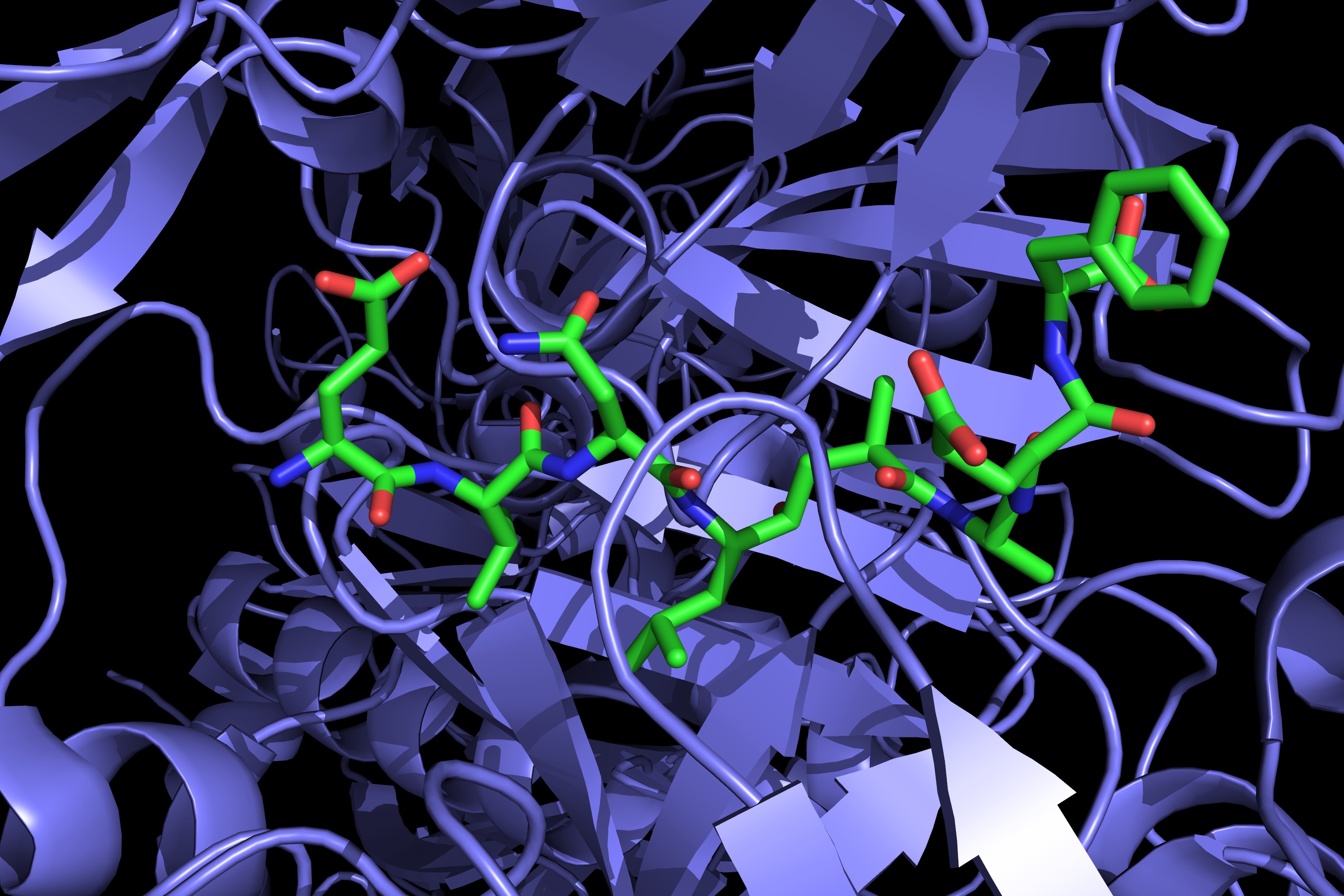
Apart from achieving blood-brain barrier penetration by BACE1 inhibitors, minimizing the inhibition of cathepsin D (CatD) is the obstacle that faces this research. CatD is an aspartyl protease with high sequence homology to BACE1 at the active site. It has been shown that the inhibition of CatD gives rise to toxic side effects. Once these two hurdles have been overcome, there will likely be a cure for the Alzheimer’s disease. Below, by the way, you can see a picture of the first report of BACE1 co-crystallized with a peptide inhibitor some 15 years ago (pdb id 1FKN).