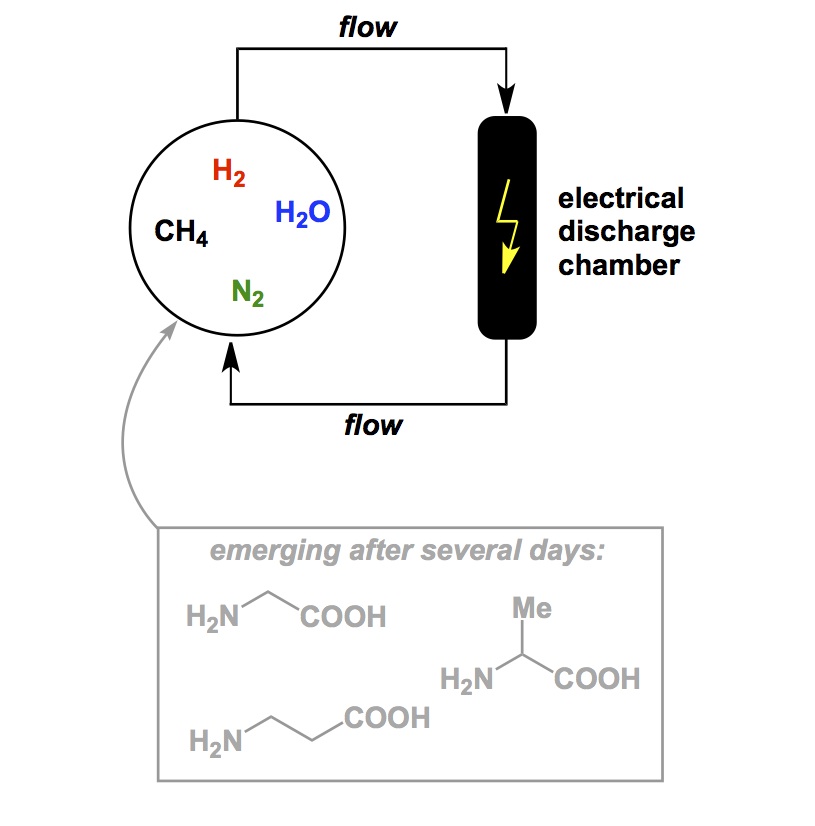
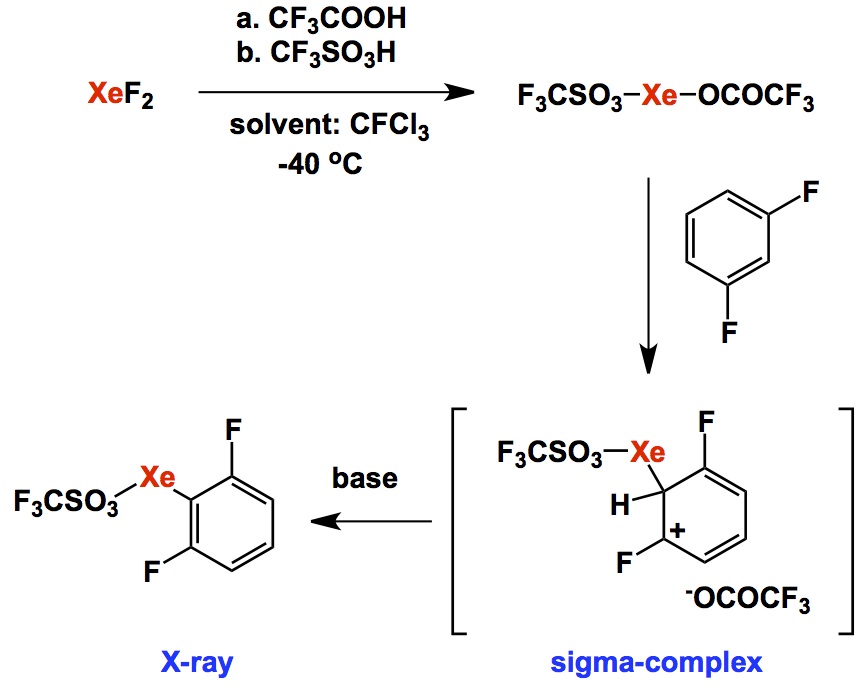
As you have probably noticed, I am not using this blog as a forum to dwell on controversial, if not scandalous, topics pertaining to research. My goal is to comment on the exciting aspects of chemistry using current literature and some of the classics from the past that may have gone unnoticed or are being forgotten. We do need to have a system for our students to keep in mind that certain areas, despite claims by some overzealous practitioners of modern synthetic chemistry, were not actually discovered in the past 5 years. Instead, they might have remained under the radar for a while. I can speak from my personal experience in the field of trifluoromethylation. When I was working on my PhD aimed at silicon-based trifluoromethylation reagents with Prakash and Olah, very few people were involved in it (apart from the tightly knit fluorine community). Nowadays, everyone and their uncle is running trifluoromethyl group transfer processes and I notice, with surprise, that Burton’s, Prakash’s, Chambers’ papers are often not cited at all. Alas, rediscovery is a common irony of chemistry.
There is a lot to be said about scientific scandals that erupt from time to time. These scandals are sometimes caused by a report of an alleged attempt to manipulate data. I don’t even want to rehash this right now – you all know some of the recent cases, I will not turn my blog into a tabloid. I do think that deliberate mispresentation of data is wrong for many reasons. But discrepancies eventually get caught and mechanisms for catching fraud are more sophisticated now compared to 20, or even 10, years ago. Publishers are becoming irritated with those in the blogging community, whose goal is to seek and disclose fraudulent research. In the opinion of many journals and their editorial boards, exposure cannot be left to bloggers because there is little accounting for what is put in the public domain using this mechanism. Chembark (http://blog.chembark.com) is an interesting example. Recently, its owner Paul Bracher wrote a long rebuttal to the editorial that appeared in ACS Nano. You can take a look at it yourself. While I agree that spreading news about research misconduct using blogs is far from an ideal mechanism, we should remember that some people’s passion is to find faults in others’ papers. This activity is going to be difficult to regulate. However, if someone equates exposure of mistakes to the advent of internet, I would direct them to Caltech’s Richard Marsh. The phrase “getting Marshed” was coined in the 80’s when Dr. Marsh would periodically publish a paper in which he would comment on the mistakes he has found in published crystallographic group assignments. I can tell you: everyone was wary of getting Marshed.