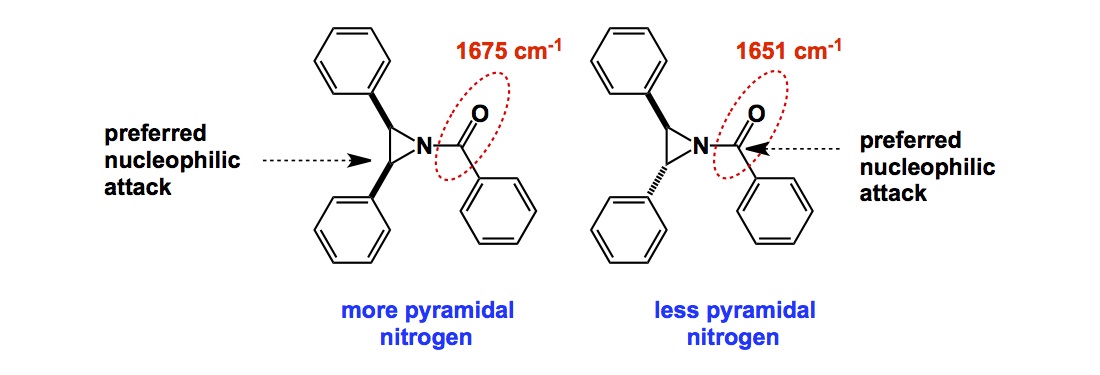
Synthetic chemists do not use a lot of math. We rely on it, there is no question about it, especially when it comes to our daily NMR experiments. Indeed, there is just so much math in the FID treatment alone. It just does not seem to affect our hypothesis-generating abilities, so we do not have a need to worry about all those complexities.
When I was doing my protein experiments with Elena Dobrovetsky during my sabbatical stay at SGC, it seemed that the “C1V1 = C2V2 (C: concentration, V: volume)” equation ruled our days. I suppose this one simple relationship is the most widely used piece of math when you need to prepare your 142nd buffer of the day.
But what is the single most important mathematical equation every organic chemist should know? In my personal view, it is the one describing the relationship between temperature, energy, and the ratio of two molecular entities expressed as K:
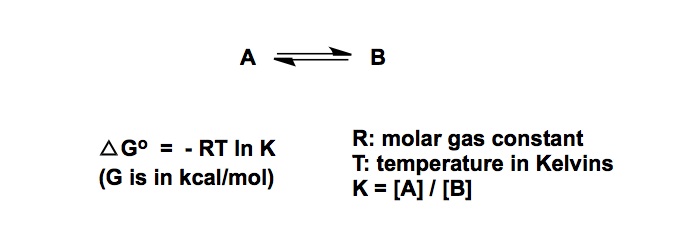
We normally refer to K as the equilibrium constant, but I wish we had a more generic term. This is because the value of the equation is in that it relates the difference in energy barriers to the ratio of products or transition states. Thus, the equation covers the domains of both thermodynamics and kinetics. Below you will see that equation again, this time rewritten in a more palatable way for quick calculations at room temperature. An immediate consequence is that each factor of 10 between the respective ratios of A and B (these could be ground or transition states) translates into a free energy change of about 1.4 kcal / mole.