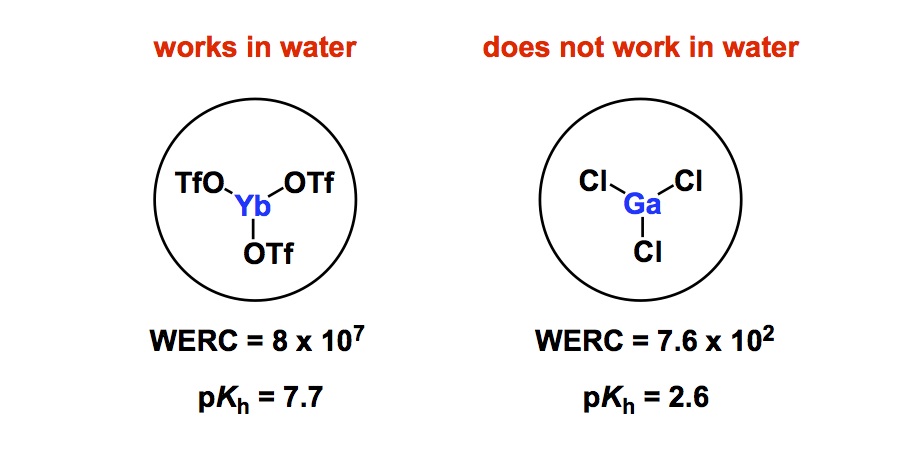
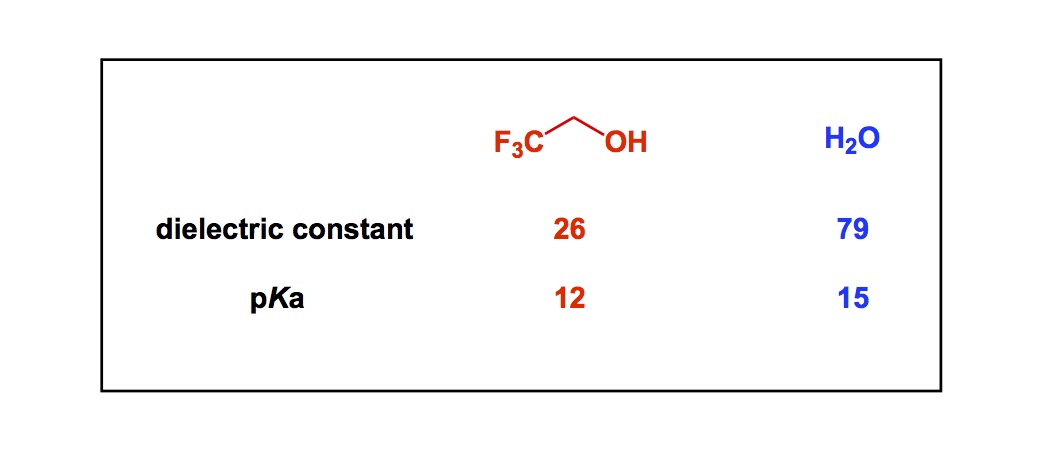
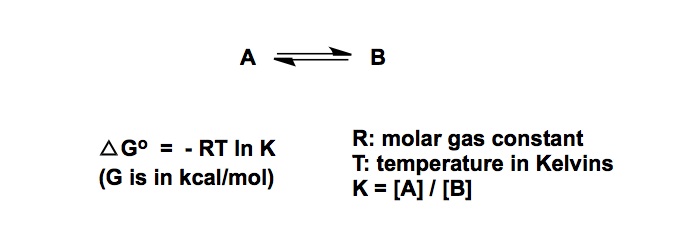
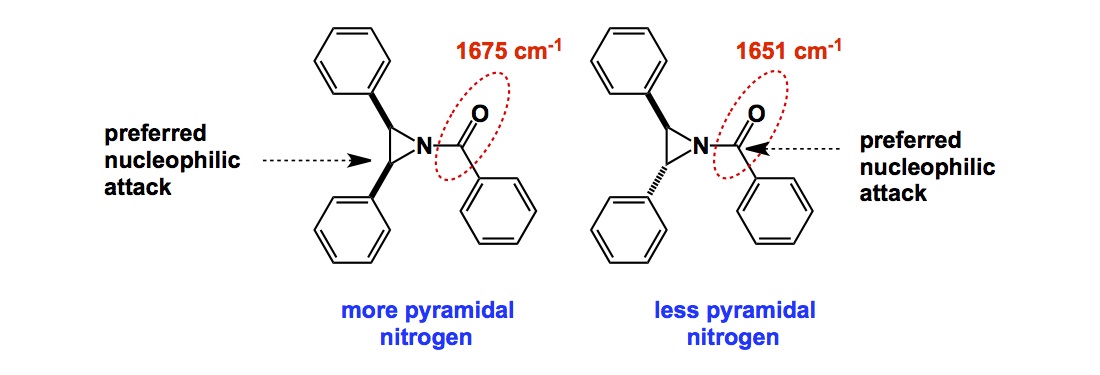
In terms of basic science, control over cis– vs trans- amide bond geometry is one of the ongoing research areas pursued by my lab. We think that this problem is important for many reasons that range from fundamental physical organic chemistry to one’s ability to dictate conformations in complex polyamide macrocycles. I have already blogged about some elements of amide cis / trans interconversion. Recently, my lab has uncovered an interesting case that points to the possibility of kinetic selection between these rotamers. On the heels of our findings, I started to think about the smallest possible ring where a clear-cut cis/trans interconversion can be observed. Below is an old and very thought-provoking paper by North and Zagotto. Apparently, amide geometry in the 8-membered ring that you see is determined by the relative configuration of the two chiral centers. Strikingly, the two cyclic diastereoisomers have different preferences for the amide bond geometry. It is highly unlikely that the cis-amide (case B) is present in the starting linear dithiol before oxidative cyclization. What likely happens in the case B is a thermodynamically controlled cyclization that involves product isomerization into a more stable (NB: in this particular instance!) cis-amide. I will leave it up to you to wonder why the cis-amide is preferred in the cyclic diastereoisomer corresponding to B. I do think that disulfide’s flexibility might be playing a role in allowing the final isomerization to take place. To my knowledge, the 8-membered ring shown below is the smallest cycle that shows such interesting cis/trans amide behavior. If you now of a smaller system, please let me know. 
http://pubs.rsc.org/en/Content/ArticleLanding/1993/C3/C39930000641#!divAbstract