I wanted to talk about something really sweet, but last Friday’s encounter with my friend and colleague Dwight Seferos has been lingering in my head over this long weekend. Dwight brought to my attention a recent Angewandte paper by Yan, Zhang and co-workers. Upon reflection, this might represent another example of not using Occam’s razor (see my previous post: https://amphoteros.com/2015/02/24/treading-lightly/). But… There are other issues here, more systemic in nature, and I would like to comment on those. The reason for this paper being “the last straw” for me is that more and more frequently I see bastardization of structures in materials’ science papers.
When we learn first year chemistry, we come to terms with a distinction between covalent and ionic bonds. Kekule’s depiction of covalent bonds has been tremendously useful and has allowed us to do everything from counting electrons to being responsible for how reasonable structures might appear in print. In contrast, ionic bonds are much less “directional”. Therein lie both advantages and disadvantages of the two types of interatomic connections. When I saw Scheme II in the aforementioned Angewandte paper, I could not believe my eyes. This depiction instantly took the air out of the work. I am not claiming to be an expert in polyazacene materials for lithium storage, but I do not need to be one in order to take offense in the way the authors rendered their structures and offered a rationale for the observed effect. I will faithfully reproduce the drawing I saw in the paper:
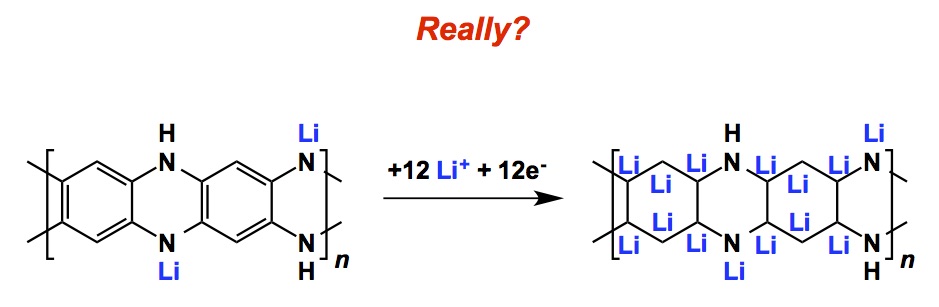
http://onlinelibrary.wiley.com/doi/10.1002/anie.201503072/abstract
These mean lithium atoms seem to do a number on the poor polyazacene molecule, don’t they? I wonder what David Collum of Cornell (http://chemistry.cornell.edu/faculty/detail.cfm?netid=dbc6) would think about this picture. He has spent his whole career painstakingly teaching us about carbon-lithium bonds…