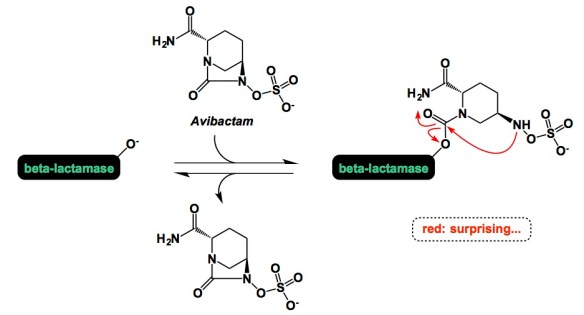
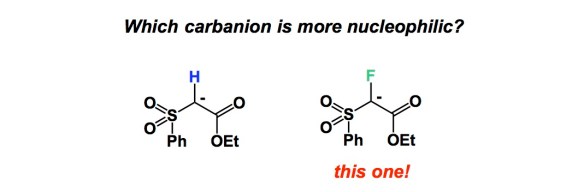
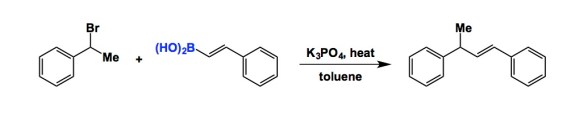
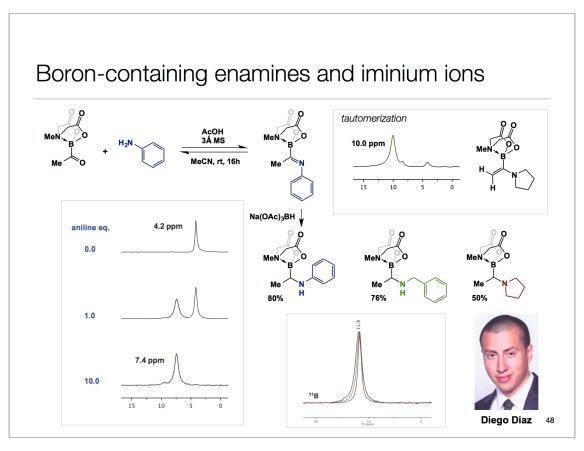
It is tempting to overemphasize the role of sophisticated equipment and fancy set-ups in modern chemistry. This is why I am encouraged when people do more with less. Of particular significance are colorimetric tests that do not involve any dyes that undergo insane conformational changes to reveal what is happening at the molecular level. Below is a simple reaction I came across while looking for examples of boron/amide interaction (something that is of interest to our lab at the moment). This is Matteson’s old work and the best part of the experimental section is the description of how the starting boronic acid was stripped off the boric acid impurity. The authors state that “… was freed from boric acid by treatment with 50 mL of methanol and distillation until the distillate showed no green boron color in the flame when ignited.” We need our students to never forget that trivial tests such as this simplify life and make practitioners of synthesis look resourceful when they present research findings at conferences or job interviews. The irony is that this can look more impressive than the seemingly more elaborate modern techniques. Let me know if you are aware of additional examples where deep insights about chemical matter are made using everyday tools.


 With this vignette, I am going to send a special hello to John, who will be missed. This area of research is now in the hands of Solomon Appavoo, a first year graduate student in my lab. Let’s see where he takes it.
With this vignette, I am going to send a special hello to John, who will be missed. This area of research is now in the hands of Solomon Appavoo, a first year graduate student in my lab. Let’s see where he takes it.