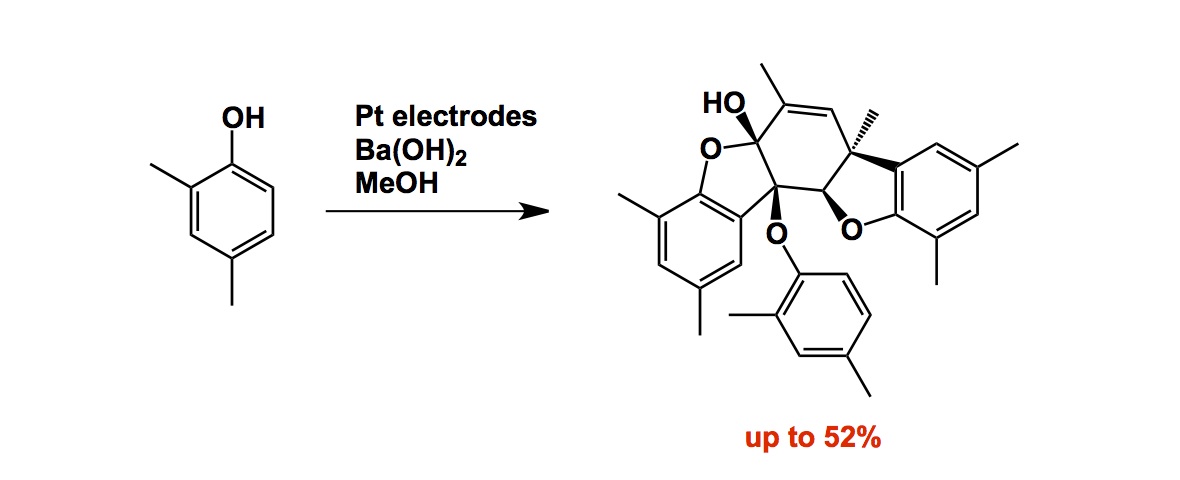
I think all students in sciences rightfully ask questions about their future employment. We live in uncertain times and job prospects are of great concern to our students. I still remember 1997 when I was a PDF at Scripps. My friends all thought I was a complete fool to go into academia. They kept saying: “rather than doing something so crazy, you need to think about what’s more or less guaranteed for life, Andrei”… They referred to jobs at Pfizer, Merck, etc. Of course you know what happened then. The ground rule that governs capitalism is the bottom line. Accordingly, it became profitable for big corporations to outsource production and, later, discovery, to places like China and India. People started losing their jobs in North America and it has been tough here for several years. But guess what? The pendulum eventually swings back. The same laws that governed leakage of jobs to Asia must eventually move to equalize the costs here and there. You might still say that the difference in costs remains large and will continue to stay that way for a while. Well then… I give you widespread corruption in places like China. We are all aware of some serious issues that keep popping up these days, all pointing to fraudulent practices in the pharmaceutical R and D in China. Several big companies are now implicated in scandals and we can foresee a situation when it becomes too risky to do business there. It is always that good old risk/benefit analysis. The jobs may then very well come back to North America. Check out the following high profile report published in Science:
Where could the jobs of the future come from?
Reply