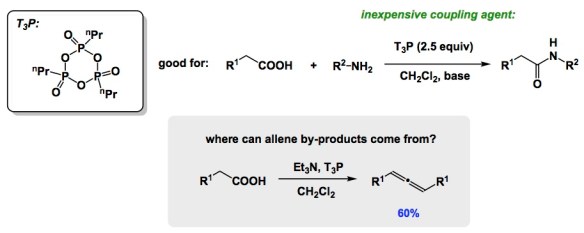
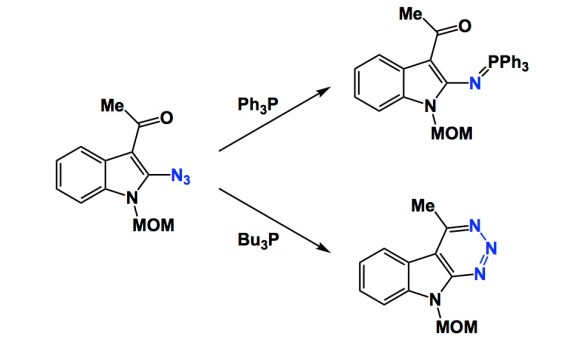
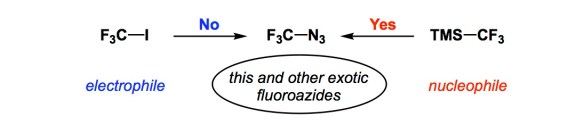
I am a fan of small amine-containing compounds with relatively short history in synthetic organic chemistry. Such molecules are admittedly hard to come by, but when I see them, I marvel at what might be done with them and why people have not considered them more broadly.
The other day I was flipping through the 2017 Strem catalog for no logical reason other than I got this shiny new booklet in the mail and felt guilty to toss it straight into the blue recycling bin, the destination of all catalogs I receive on a weekly basis. My attention got piqued by 2-aminoethane-1,1-disulfonic acid (let’s call it ADSA), which is offered by Strem for some unknown reason (metal catalysts is their main focus). Unaware of ADSA’s existence, I looked through standard search engines and found very little prior to 2010. There was some work done by Wagner and co-workers in the 60’s, but not much since. The synthesis of this compound is simple, yet interesting as it involves a modified Ritter reaction with oleum, decarboxylation, and sulfonation of the enamide. ADSA offers as an outstanding way to improve aqueous solubility of fairly hydrophobic molecules such as Alexa Fluor dyes. I find the geminal bis(sulfonate) functionality rather interesting because it reminds me of bis(phosphonates), which are of course miles ahead in terms of demonstrated use and significance as components of drugs that prevent the loss of bone mass.

http://www.sciencedirect.com/science/article/pii/S0040403910006908