Humans have celebrated food since the dawn of civilization. We are what we eat. For centuries, painters have been drawing inspiration from all manner of cooked and uncooked specimens. Here is Jan Davidszoon de Heem’s “Still Life with Fruit and Ham” from 1648.

Today I have my PhD student Ben Chung as a guest on this blog. Ben gave an inspiring talk about chemistry of cooking last week. Here is what he wrote on the subject of heterocycles your mother wants you to eat…
Ben:
“Heterocycles are cyclic compounds with one or more elements other than carbon within their ring structure. One of the earliest examples of heterocycle synthesis in the lab was the isolation of alloxan through uric acid oxidation by Brugnatelli back in 1818. However, humans have been making heterocyclic compounds long before then… by cooking food! Through my adventures in learning about food chemistry and molecular gastronomy, I’ve stumbled upon many interesting molecules that are formed through cooking. These molecules are mainly produced by the Maillard reaction, which describes the reactions that occur between sugars and amino acids upon heating.
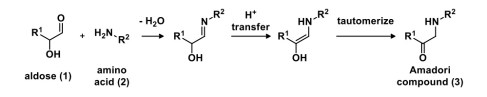
Using a generic aldose (1) as an example, the amino group of an amino acid (2) reacts with the aldehyde to generate N-glycosides, which then undergo an Amadori rearrangement to generate an amino-functionalized ketose, known as the Amadori compound (3). The net transformation is N-functionalization of the aldose as well as transfer of the carbonyl oxidation state from the terminal end to the middle of the molecule.
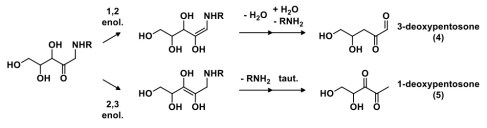
The Amadori compound then undergoes additional transformations, generating a class of compounds known as deoxyosones (e.g., deoxypentosones 4 and 5). The regioselectivity of the enolization determines the type of deoxyosone that’s formed, and deoxyosones are named by the chain length and the carbon number at which deoxygenation has occurred.

It is these deoxyosone intermediates that can be further dehydrated, cyclized, and functionalized by ammonia, hydrogen disulfide or methanethiol (derived from amino acid degradation pathways) to generate a large variety of heterocyclic flavour molecules. An example of 3-deoxypentosone degradation is shown above; analogous pathways can occur for 1-deoxypentosones as well. The structures of a few heterocyclic molecules derived from the Maillard reaction are shown below, as well as the aromas they have.

Of course, the variety of different sugars and amino acids present in any given sample of food means that, following Maillard reaction, it is possible to achieve an almost infinite combination of aromas and flavours. Who would’ve guessed that chefs possessed such mastery over heterocyclic synthesis?”
Here is a good read:
http://pubs.acs.org/doi/abs/10.1021/ar8002078